易基因:全基因组DNA甲基化和小RNA分析揭示甘蓝型油菜种子的基因组不对称性
Posted E-GENE
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了易基因:全基因组DNA甲基化和小RNA分析揭示甘蓝型油菜种子的基因组不对称性相关的知识,希望对你有一定的参考价值。
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
多倍体是被子植物基因组进化中的一种持续现象,有助于现存开花植物的多样性。甘蓝型油菜(Brassica napus)是世界上最重要的被子植物油料作物品种之一,起源于Brassica rapa(An)和Brassica oleracea(Cn)的种间杂交。尽管转录组学中基因组优势的趋势开始显现,但多倍体在生殖发育过程中的表观遗传学和小RNA(small RNA,sRNA)调控机制知之甚少。种子是向新孢子体代的关键发育过渡,并随时间发生表观遗传修饰。双二倍体(amphidiploids)种子发育的协调需要祖细胞(Progenitor cell)基因组的表观遗传平衡。虽然DNA甲基化和RNA干扰是支持发育的关键过程,但将它们与种子发育联系起来的许多细节仍在出现,尤其是在异源多倍体中。
2023年05月17日,加拿大曼尼托巴大学生物科学系Mark F Belmonte团队在《The Plant Journal》杂志发表题为“Genomic asymmetry of the Brassica napus seed: epigenetic contributions of DNA methylation and small RNAs to subgenome bias”的研究论文,该研究利用WGBS和sRNA-seq等实验揭示了DNA甲基化和小RNA在甘蓝型油菜种子亚基因组偏好中的表观遗传作用。

标题:Genomic asymmetry of the Brassica napus seed: epigenetic contributions of DNA methylation and small RNAs to subgenome bias(甘蓝型油菜种子的基因组不对称性:DNA甲基化和小RNA对亚基因组偏好的表观遗传贡献)
时间:2023-05-17
期刊:The Plant Journal
影响因子:IF 7.091
技术平台:WGBS、sRNA-seq等
研究摘要
本研究分析了甘蓝型油菜(B. napus)种子发育过程中,An和Cn两个亚基因组以及祖代缩减的基因组(ancestral fractionated genome)中DNA甲基化和小干扰RNA(small interfering RNA,siRNA)图谱的偏好发生率。研究结果表明,Cn亚基因组的siRNA表达和胞嘧啶甲基化的普遍存在偏好,其中Cn亚基因组启动子区域的DNA甲基化尤为富集。此外甘蓝型油菜祖代三倍体亚基因组中的siRNA转录模式是保守的,而在An和Cn两个亚基因组中不保守。从基因组缩减和多倍体化角度研究了甘蓝型油菜种子的甲基化模式与基因、启动子区域、siRNA位点和转座元件(transposable element,TE)相关。本研究为甘蓝型油菜种子发育过程中选择性沉默Cn亚基因组的表观遗传调控提供了证据,并探讨了基因组缩减对甘蓝型油菜种子表观遗传组分的影响。
图1:甘蓝型油菜种子发育的表观遗传模型假设
- 从配子体发生(gametogenesis)到成熟期结束,种子发育可分为5个离散时期:胚珠期(ovule,OV)、球形期(globular,GLOB)、心形期(heart)、绿熟期(mature green,MG)和干种子期(dry seed,DS)。所有五个时期进行sRNA-seq测序分析,同时对球形期(GLOB)和绿熟期(MG)的种子进行全基因组重亚硫酸盐测序(WGBS),GLOB期和MG期分别代表了形态发生和成熟开始的关键发育转变。
- 共线性图显示了双二倍体甘蓝型油菜基因组中每条染色体最大的连续共线性区,Brassica rapa(Br)的An亚基因组,Brassica oleracea(Bo)的Cn亚基因组。
- 表观遗传保守性的假设模型——基因组的broad共线区表明两个亚基因组之间的高度保守。这些区域的保守可能导致生殖中类似的表观遗传结构。
研究结果
(1)甘蓝型油菜种子甲基化组单碱基分辨率图谱揭示了Cn亚基因组偏好
表1:甘蓝型油菜叶片、GLOB期和MG期种子基因组上1 kb窗口的甲基化百分比
图2:甘蓝型油菜种子发育中的DNA甲基化
- 平均胞嘧啶甲基化水平。显示球形期(GLOB,绿色)、绿熟期(MG,蓝色)和质体(plastidial,灰色)基因组所有覆盖胞嘧啶的平均甲基化。
- 甘蓝型油菜An和Cn两个亚基因组中甲基化缩减的基因组水平:最少缩减(LF)、最多缩减(MF1和MF2)。分位数箱线图显示在CG、CHG和CHH中,叶片、GLOB期种子和MG期种子的共线性区域1 kB窗口中的胞嘧啶甲基化水平。
(2)启动子元件的差异甲基化靶向参与甘蓝型油菜种子碳代谢和发育的基因
图3:在种子发育全过程中,不同基因组元件的DNA甲基化变化。
- 百分比堆积柱形图显示球形期(GLOB)种子和绿熟期(MG)种子以及叶片之间的高甲基化和低甲基化基因(GENE)、1 kb上游调控区(启动子,PROM)和转座元件(transposable element,TE)。分别为GLOB期种子(橙色)或MG期种子(浅橙色)中的高甲基化,GLOB期种子(黄色)或MG期种子(浅黄色)中的低甲基化,GLOB期种子和MG期种子中的高甲基化(深蓝)或低甲基化(浅蓝)。
- 种子发育GLOB期和MG期的启动子差异甲基化基因GO富集分析。深色表示高度富集。
(3)相对于其他蛋白质编码基因,转录因子(TF)低甲基化
表2:RE(TE)、基因体和启动子元件(TSS上游1 kB窗口)、TF基因体和启动子以及siRNA簇的甲基化百分比
(4)缩减的基因组在基因和启动子上表现出不同的甲基化
图4:基因组的DNA甲基化表征:基因、启动子区域(prom)、重复元件(RE)和siRNA位点。
以甘蓝型油菜(Brassica napus)的缩减基因组(最少缩减(LF)、最多缩减(MF1、MF2))及其对应的亚基因组(A、C)划分的箱型图和晶须图。
(5)种子成熟以转录的siRNA位点数量增加为表征
图5:甘蓝型油菜种子发育过程中的小干扰RNA(siRNA)reads积累
- 种子发育不同时期的文库中总siRNA位点的reads/M(RPM)比例表现出出一致的Cn亚基因组偏好。
- 在种子发育早期,较少siRNA位点占总reads较大比例,而reads在绿熟期(MG)比胚珠期(OV)、球形期(GLOB)、心形期(heart)分布更均匀。
(6)甘蓝型油菜种子中转座元件(TE)和基因启动子区域编码大部分转录sRNA
图6:由转座元件(TE)、基因体和启动子编码的小干扰RNA(siRNA)簇。
- siRNA在种子发育全过程中以一致的Cn亚基因组偏好性积累。
- 源于基因体的siRNA在DS期积累最强,数量少于由启动子区编码的siRNA。
- 启动子区siRNA在绿熟期(MG)表达最为富集,从胚珠期(OV)到MG期表达增加。启动子区域比基因体编码的siRNA表现出更强的亚基因组偏好。
- 对与启动子区域相关的编码基因(每个种子阶段最高表达的500个siRNA簇)进行GO富集分析,该启动子区域编码每个种子阶段最高表达的500个siRNA簇。
(7)祖代基因组缩减对种子发育过程中的siRNA积累影响不大
图7:种植发育OV期、GLOB期、heart期、MG期和DS期的最少缩减(LF)和最多缩减(MF1和MF2)基因组>1 read/M(RPM)siRNA位点小提琴图
(8)基因组缩减对非蛋白编码元件的影响小于对基因体和启动子区域的影响
图8:在CHG和CHH甲基化环境中,基因、启动子、转座元件(TE)和小干扰(si)RNA位点的高度不显著比较(P>0.1,Mann-Whitney-Wilcoxon)。
(a)球形期(GLOB)、(b)绿熟期(MG)、(c)GLOB-MG的比较。
结论
总之,本研究为甘蓝型油菜种子中Cn亚基因组的表观遗传偏好和基因组中非蛋白编码元件的表观遗传调控机制提供了证据。本研究数据表明,基因组缩减并没有实质性地改变转座子和种子发育过程中calling的siRNA甲基化谱。种子的表观遗传结构可能是决定甘蓝型油菜进化轨迹的重要因素,尤其与多倍体作物相关。
关于易基因全基因组重亚硫酸盐测序(WGBS)技术
全基因组重亚硫酸盐甲基化测序(WGBS)可以在全基因组范围内精确的检测所有单个胞嘧啶碱基(C碱基)的甲基化水平,是DNA甲基化研究的金标准。WGBS能为基因组DNA甲基化时空特异性修饰的研究提供重要技术支持,能广泛应用在个体发育、衰老和疾病等生命过程的机制研究中,也是各物种甲基化图谱研究的首选方法。
易基因提供的全基因组甲基化测序技术通过T4-DNA连接酶,在超声波打断基因组DNA片段的两端连接接头序列,连接产物通过重亚硫酸盐处理将未甲基化修饰的胞嘧啶C转变为尿嘧啶U,进而通过接头序列介导的 PCR 技术将尿嘧啶U转变为胸腺嘧啶T。
应用方向:
WGBS广泛用于各种物种,要求全基因组扫描(不错过关键位点)
- 全基因组甲基化图谱课题
- 标志物筛选课题
- 小规模研究课题
技术优势:
- 应用范围广:适用于所有参考基因组已知物种的甲基化研究;
- 全基因组覆盖:最大限度地获取完整的全基因组甲基化信息,精确绘制甲基化图谱;
- 单碱基分辨率:可精确分析每一个C碱基的甲基化状态。
易基因科技提供全面的DNA甲基化研究整体解决方案。
参考文献:
Ziegler DJ, Khan D, Pulgar-Vidal N, Parkin IAP, Robinson SJ, Belmonte MF. Genomic asymmetry of the Brassica napus seed: epigenetic contributions of DNA methylation and small RNAs to subgenome bias. Plant J. 2023 May 17. doi: 10.1111/tpj.16254. PubMed PMID: 37195091.
相关阅读:
大豆:多组学揭示体细胞胚胎发生过程DNA甲基化与发育转变的关系
易基因:全基因组ChIP-seq分析揭示细菌转录因子PhoB的基因内结合位点|mBio
大家好,这里是专注表观组学十余年,领跑多组学科研服务的易基因。
细菌编码许多转录因子(transcription factor,TF),这些转录因子通过与启动子周围的DNA结合并调控RNA聚合酶(RNAP)全酶以结合启动子DNA或异构化为主动转录构象的能力来调节转录起始。目前对TF功能的研究几乎集中在调节基因上游基因间区域的TF结合位点。然而,基因组规模分析鉴定出大量的TF结合位点位于基因内(within genes),基因内TF结合位点的比例在不同TF之间具有显著差异。尽管基因内存在大量TF结合位点,但对其功能知之甚少。
大肠杆菌(Escherichia coli)TF PhoB是一种保守的转录因子,可调控参与磷酸盐稳态的基因转录。大部分PhoB结合位点位于基因内,这些基因内结合位点与可检测的转录调控无关,且在进化上不保守。许多基因内PhoB位点位于H-NS结合区域,可能是由于PhoB和H-NS的共同序列偏好。
2023年04月17日,《mBio》杂志发表了题为“Genome-Wide Mapping of the Escherichia coli PhoB Regulon Reveals Many Transcriptionally Inert, Intragenic Binding Sites”的研究论文,该研究通过对细菌转录因子PhoB结合位点的ChIP-seq、RNA-seq整合分析,揭示了大肠杆菌PhoB的许多基因内转录因子结合位点和转录调控机制。
标题:Genome-Wide Mapping of the Escherichia coli PhoB Regulon Reveals Many Transcriptionally Inert, Intragenic Binding Sites.(大肠杆菌PhoB调控元件的全基因组定位揭示了许多转录惰性的基因内结合位点)
时间:2023-04-17
期刊:mBio
影响因子:IF 7.786
技术平台:ChIP-seq、RNA-seq、RT-qPCR等
研究摘要:
基因组规模分析揭示许多转录因子结合位点在基因内部,引发了关于这些结合位点功能的问题。最近研究揭示细菌基因内的大量转录因子结合位点,但绝大多数结合位点的功能尚未研究。本研究绘制了转录因子PhoB在大肠杆菌基因组中的结合,揭示了大多数PhoB结合位点都位于基因内。分析结果表明,基因内PhoB结合位点与重叠基因调控无关。数据揭示了细菌可以耐受大量非调控性基因内转录因子结合位点的存在,且这些结合位点不受选择性压力影响。
研究使用染色质免疫沉淀测序(ChIP-seq)和转录组测序(RNA-seq)对pho调控元件进行高分辨率的全基因组分析。研究对一组已知的pho调控基因进行分析和扩展,并鉴定出许多基因内PhoB结合位点。分析结果显示,绝大多数基因内PhoB结合位点不保守,且与可检测的调控功能无关。本研究数据表明,单个基因内PhoB位点无功能,转录因子可以结合许多基因内位点,对局部转录(local transcription)几乎没有影响。
实验结果:
(1)磷酸盐限制条件下PhoB的全基因组结合
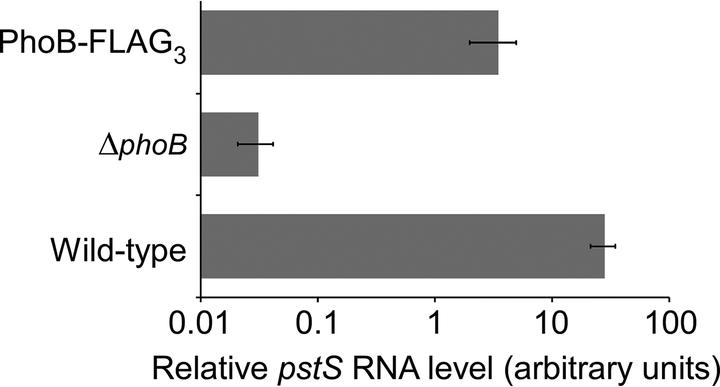
图1:C末端带有FLAG3标签的PhoB活性部分降低。qRT-PCR检测在低磷酸盐条件下生长的细胞中,野生型MG1655/pBAD24(wild type)、MG1655 ΔphoB(CDS091)/pBAD24(ΔphoB)和MG1655 phoB-FLAG3(DMF34)/pBAD24(phoB-FLAG3)中相对minD RNA对照的pstS RNA水平。值是三个生物学重复平均值;误差线表示±1个标准差。

图2:ChIP-seq鉴定PhoB结合位点。
- ChIP-seq数据:(i)低磷酸盐条件下的无标签对照,(ii)低磷酸盐条件下的PhoB-FLAG3标签,(iii)高磷酸盐条件下的PhoB-FLAG3标签。三个基因组区域,其中一个数据来自两个生物学重复。x轴表示基因组位点。y轴表示归一化序列reads覆盖范围,正值表示序列reads比对到正义链,负值表示序列reads比对到反义链。
- 每个ChIP-seq peaks周围100 bp区域显著富集的DNA序列motif。
- PhoB结合位点相对于ChIP-seq peaks中心区域位点分析。
- ChIP-seq鉴定的PhoB结合位点的基因组背景饼图。“基因内和上游”位点是基因内但注释基因起始上游<200bp位点。
表1:ChIP-seq鉴定的PhoB结合区域列表

图3:ChIP-seq和ChIP-ChIP数据集比较分析
- 本研究ChIP-seq数据和已发表的ChIP-ChIP数据集之间的共有区域中,每个ChIP-seq peaks周围100 bp区域显著富集的DNA序列motif。
- 本研究ChIP-seq数据的特异性区域,每个ChIP-seq peaks周围100 bp区域显著富集的DNA序列motif。
(2)pho调控元件的再评估

图4:大肠杆菌野生型和ΔphoB菌株的RNA-seq分析。散点图显示野生型(MG1655/pBAD24)或ΔphoB(CDS091/pBAD24)细胞中所有基因的相对RNA水平。每个数据点对应一个基因,三角形数据点代表先前报道的pho调控元件中的基因,红色三角形表示该转录本ChIP-seq鉴定的上游PhoB位点,灰色三角形表示没有上游位点。红色圆圈表示以前没有报道过的基因位于pho调控元件中,但在ChIP-seq中鉴定出上游PhoB位点。蓝色圆圈表示ChIP-seq鉴定出的内部PhoB位点基因。灰色圆圈表示所有其他基因。
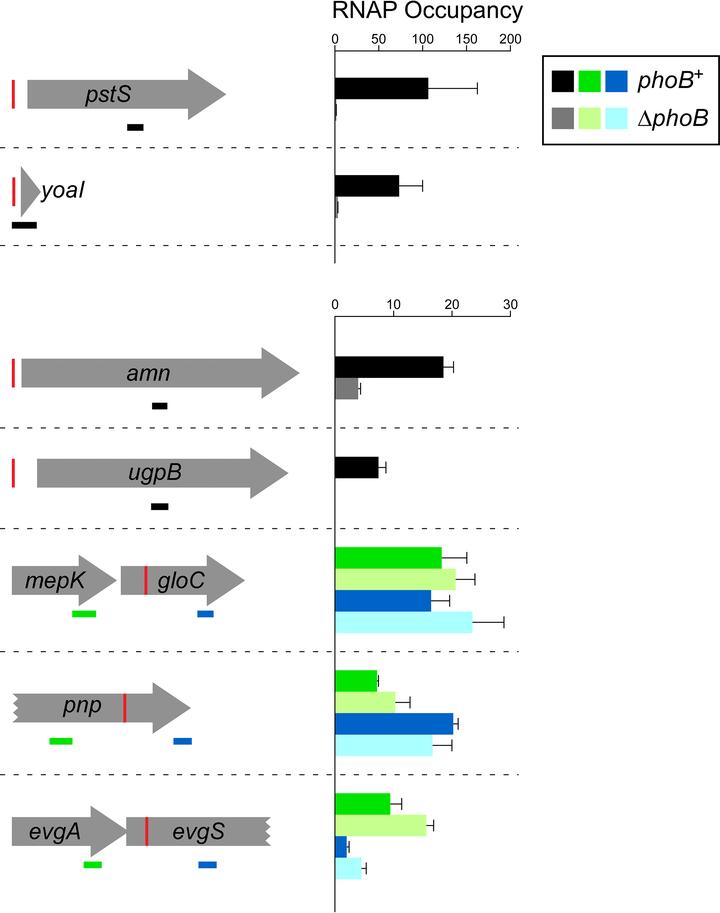
图5:pho调控元件潜在成员基因中RNAP(β亚基)占有率差异。
野生型MG1655(深色条)和MG1655 ΔphoB(CDS091;浅色条)中,ChIP-qPCR检测的RNAP(β亚基)占有率是pho调控元件潜在成员的基因内区域。左侧示意图显示上游或内部PhoB位点的基因(红色垂直线)。水平条表示ChIP-qPCR中PCR扩增子位点,黑色条表示PhoB位点上游基因内的扩增子,绿色条表示基因内PhoB位点上游的扩增子,蓝色条表示基因内PhoB位点下游的扩增子。
(3)起始RNAP的PhoB依赖性招募

图6:野生型和ΔPhoB细胞PhoB结合位点周围σ70占有率差异。
散点图显示ChIP-seq鉴定的phoB结合位点周围400bp区域在野生型MG1655和MG1655 ΔphoB(DMF84)中的归一化σ70占有率。每个数据点代表一个PhoB结合位点。基因间结合位点由红色数据点表示,基因内结合位点由蓝色数据点表示。与PhoB结合位点相关的基因在野生型和ΔPhoB细胞的σ70占有率相差>2倍。
(4)H-NS与许多基因内PhoB位点相关联,但不阻断RNAP招募

图7:H-NS抑制许多启动子的转录
- 通过ChIP-seq鉴定的野生型MG1655和MG1655 Δhns(AMD565a)PhoB结合位点周围400 bp区域的归一化σ70占有率散点图。
- 通过ChIP-seq检测MG1655Δhns(AMD565a)细胞中所有σ70结合位点,鉴定出野生型MG1655和MG1655 Δhns(AMD565a)的归一化化σ70占有率散点图。

图8:H-NS不抑制PhoB依赖性起始RNA聚合酶招募效应
(5)PhoB结合位点的序列保守性
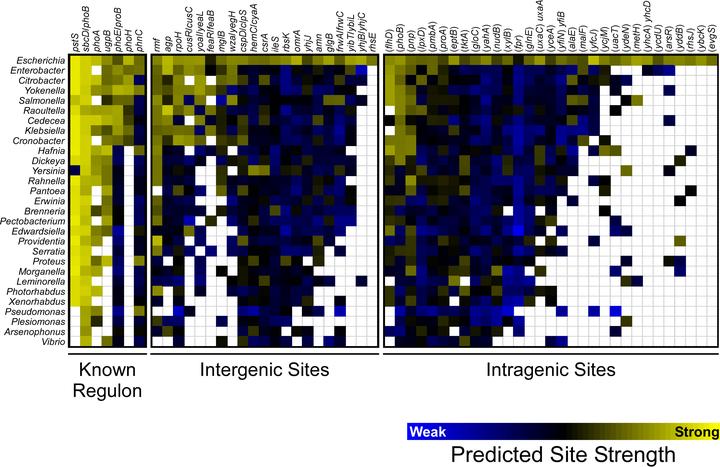
图9:PhoB结合位点在γ -变形菌属(Gammaproteobacteria)物种中的保守性
关于易基因染色质免疫共沉淀测序 (ChIP-seq)
染色质免疫共沉淀(Chromatin Immunoprecipitation,ChIP),是研究体内蛋白质与DNA相互作用的经典方法。将ChIP与高通量测序技术相结合的ChIP-Seq技术,可在全基因组范围对特定蛋白的DNA结合位点进行高效而准确的筛选与鉴定,为研究的深入开展打下基础。
DNA与蛋白质的相互作用与基因的转录、染色质的空间构型和构象密切相关。运用组蛋白特定修饰的特异性抗体或DNA结合蛋白或转录因子特异性抗体富集与其结合的DNA片段,并进行纯化和文库构建,然后进行高通量测序,通过将获得的数据与参考基因组精确比对,研究人员可获得全基因组范围内某种修饰类型的特定组蛋白或转录因子与基因组DNA序列之间的关系,也可对多个样品进行差异比较。
应用方向:
ChIP 用来在空间上和时间上不同蛋白沿基因或基因组定位
- 转录因子和辅因子结合作用
- 复制因子和 DNA 修复蛋白
- 组蛋白修饰和变异组蛋白
技术优势:
- 物种范围广:细胞、动物组织、植物组织、细菌微生物多物种富集经验;
- 微量建库:只需5ng以上免疫沉淀后的DNA,即可展开测序分析;
- 方案灵活:根据不同的项目需求,选择不同的组蛋白修饰特异性抗体。
技术路线:
易基因提供全面的DNA与蛋白互作测序方案。
参考文献:
Fitzgerald DM, Stringer AM, Smith C, Lapierre P, Wade JT. Genome-Wide Mapping of the Escherichia coli PhoB Regulon Reveals Many Transcriptionally Inert, Intragenic Binding Sites. mBio. 2023 Apr 17:e0253522.
相关阅读:
干货系列:染色质免疫共沉淀测序(ChIP-seq)的数据挖掘思路
干货系列:高通量测序后的下游实验验证方法——ChIP-seq篇
以上是关于易基因:全基因组DNA甲基化和小RNA分析揭示甘蓝型油菜种子的基因组不对称性的主要内容,如果未能解决你的问题,请参考以下文章