CMP分析流程
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了CMP分析流程相关的知识,希望对你有一定的参考价值。
参考技术A**(1)在xx范围内含有至少2个CG;(2)reads1 barcode区域的质量值控制;(3)barcode一样的reads仅保留一个 **
(1)在xx范围内含有至少2个CG
(2)reads1 barcode区域的质量值控制
TTTCCCTACACGACGCTCTTCCGATCTHHHHHHHHCGCH
TTTCCCTACACGACGCTCTTCCGATCTHHHHHHHCGHCH
TTTCCCTACACGACGCTCTTCCGATCTHHHHHHCGHHCH
TTTCCCTACACGACGCTCTTCCGATCTHHHHHCGHHHCH
(3)、barcode一样的reads仅保留一个
重复1.5、noch_arra 过程,输入文件为$s.5bp.cgcgmat.gz
s07.Pcgibed:$s.5bp.cgcgmat.rmd.gz s07.Tcgibed:$s.5bp.cgcgmat.gz T- s07.Tcgibed/$s.cgcgmat P- s07.Pcgibed/$s.cgcgmat.qc uPnorm- s07.Pcgibed/$s.cgcgmat.qc*(qc-dCGI-$s/rmd-dCGI-$s)
uP-xxx:对于血浆样本,在MePM基础上乘以 qc-dCGI-$s/rmd-dCGI-$s
P和T的差别在于P算的UMI,T算的MePM
fmg9_m.clean:
clean reads 条数
fmg9_m.qc5.clean:
对clean reads再次做前5bp的qc后的reads
fmg9_m.filter: 去掉前6bp后12bp后剩余的reads数
fmg9_m.rmfilter: 去掉含有3个及以上nonCG的reads
fmg9_m.unique_mapping: bismark mapping到基因组
fmg9_m.cgcgmat: 在-3~+3bp(含3bp)(mapping位点为0)中至少有2个CG
fmg9_m.cgcgmat.qc: 在fmg9_m.cgcgmat的基础上,reads的前5bp做过qc
fmg9_m.cgcgmat.qc.rmd: 用UMI去掉PCR重复序列
P-CGI-fmg9_m: 用UMI去掉PCR重复后落在CGI中的序列
T-CGI-fmg9_m: 不用UMI去掉PCR重复后落在CGI中的序列
qc-dCGI-fmg9_m: qc前5bp,落在dCGI区域中的reads(dCGI有3024个,分别是什么
呢?)
qc-dCGI2-fmg9_m: qc前5bp,落在dCGI2区域中的reads(dCGI2有9513个,分别是
什么呢?)
rmd-dCGI-fmg9_m: qc前5bp,去掉UMI,落在dCGI中的reads
rmd-dCGI2-fmg9_m: qc前5bp,去掉UMI,落在dCGI2中的reads
filter2_nonCGfmg9_m: 1-("total methylated C in CHG"+"total methylated
C in CHH" )/("total methylated C in CHG"+"total methylated C in
CHH" +"total C to T conversions in CHG context"+"Total C to T
conversions in CHH context"))完成filter而未去掉含3个及以上nonCG的第二端序列然后bismark比对结果
filter1_nonCGfmg9_m : 同上,第一端序列
不光用MePM衡量甲基化程度,还用测到reads含有的甲基化位点的C/C+T来衡量。
一个小问题:是否应该同时考虑reads1和reads2的信息,为了解决这个问题,
应该计算reads1和reads2重叠区域是否很多,如果基本上重复,那么reads1和
reads2的信息是一致的,仅需要考虑一条reads即可,如果reads1和reads2重叠
区域少,那么应该同时考虑两条reads的情况。这样计算有点麻烦,因为不能分别
把reads1和reads2的C加起来,C+T加起来,然后C/C+T,原因是这样会导致重叠
区域权重增大,应该是上述的C-重叠区域C,上述C+T-重叠区域C+T,然后C/C+T
才是真的甲基化程度。所以我觉得考虑一条reads足以。
正链序列(起始点)落在正链cgcgcgg上,负链序列(起始点)落在负链cgcgcgg上
问:为什么要对单碱基数据也做normalise?
答:文老师发现一个肝癌数据中C特别高,但是癌症程度并不算太高,而是由于测序深度太深造成的。那么如果只关注C的绝对值,测序越深,C的绝对值就会越高。如果测饱和了(每个阳性位点都测到了),C的绝对值不会因为测序而升高(去掉PCR duplicate后),没有测饱和的时候,用绝对值计算是要受到测序深度影响的。另外,两个病人释放不同量的ccfDNA,而其中癌症相关的都是一条,因为取血量一样,都是5ml,那么癌症相关DNA浓度一样,但是测序得到的结果(同样测序深度)就不一样了,解决办法:饱和程度。测饱和可以解决以上两个问题。
问:为什么不用CGI-qc/CGI-rmd作为duplication rate,既然T和upnor的方法本质上是一样的,upnor的优势是什么?
答:upnor的duplication rate是一样的,而去重的时候,不可能每个位点去掉重复的比例一致,只要是乘以一个固定的duplication rate,T带来的随机性就被去掉了,至于能否用CGI-qc/rmd-CGI作为duplication rate,也要筛选那些低拷贝的地方吧,不筛选得到的duplication rate,高拷贝的地方占权重会大。
1. read1_2_filter_adapter.pl
2.rm_firstx_leny.pl
3.ch3deleate.pl
4.s05.noch_arrange
5.extractCGx2.pl
6.qc5bp.pl
7.rmduppcrv2.pl
8、CGIcgcgcggv2.sh
8086汇编 cmp 指令
8086汇编 cmp 指令
cmp 是比较指令,功能相当于减法指令,只是不保存结果。
cmp 指令执行后,将对标志寄存器产生影响。
格式:cmp 操作对象1,操作对象2
功能:计算操作对象1–操作对象2
原理:通过做减法运算影响标志寄存器,标志寄存器的相关位的取值,体现比较的结果。
cmp 指令说明
一、应用使用
其他相关指令通过识别这些被影响的标志寄存器位来得知比较结果。
应用方法:用标志寄存器值,确定比较结果。

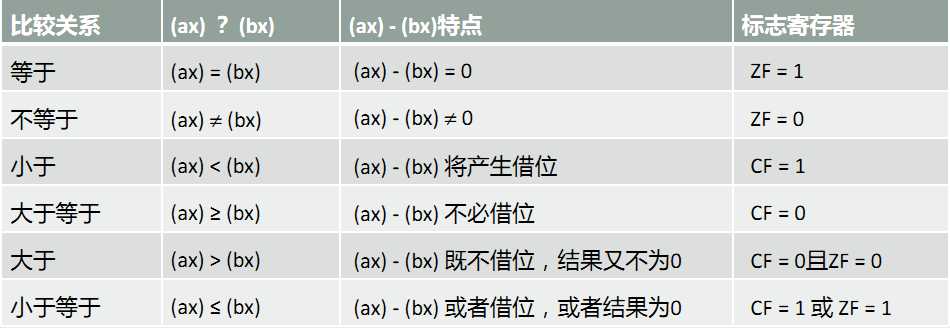
二、无符号数比较与标志位取值
思路:通过cmp 指令执行后相关标志位的值,可以看出比较的结果
指令:cmp ax,bx
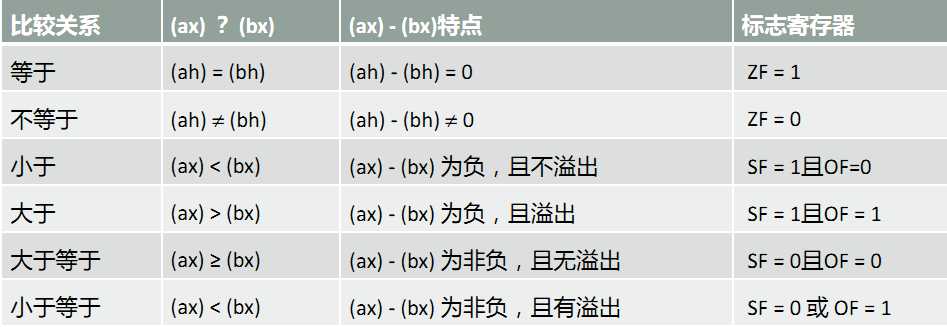
三、有符号数比较与标志位取值
问题:用cmp来进行有符号数比较时,CPU用哪些标志位对比较结果进行记录
仅凭结果正负(SF)无法得出结论,需要配合是否溢出(OF)得到结论。
示例指令:cmp ah,bh
条件转移指令
;或者其他影响标志寄存器的指令
cmp oper1, oper2
jxxx 标号
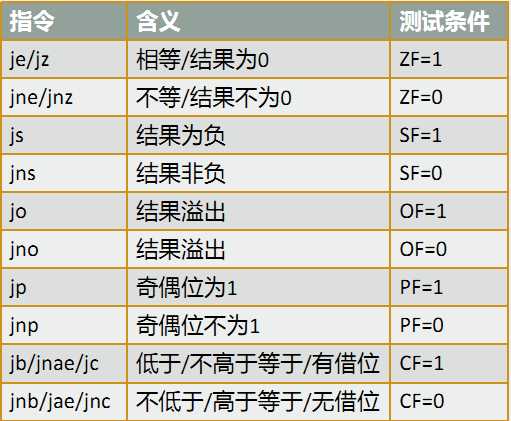
一、根据单个标志位转移的指令
二、根据无符号数比较结果进行转移的指令
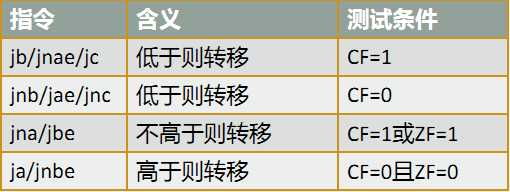
三、根据有符号数比较结果进行转移的指令
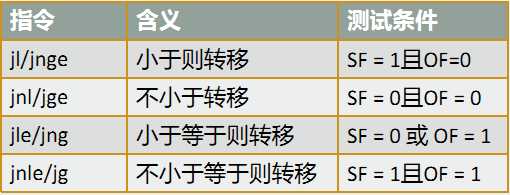
四、转移指令全写
j-Jump e-Equal n-Not b-Below a-Above L-less g-Greater s-Sign C-carry p-Parity o-Overflow z-Zero
条件准转移指令使用
jxxx系列指令和cmp指令配合,构造条 件转移指令
- 不必再考虑cmp指令对相关标志位的影响和jxxx指令对相关标志位的检测
- 可以直接考虑cmp和jxxx指令配合使用时表现出来的逻辑含义。
- jxxx系列指令和cmp指令配合实现高级语言中if语句的功能
例1:如果(ah)=(bh),则(ah)=(ah)+(ah),否则(ah)=(ah)+(bh)

例2:如果(ax)=0,则(ax)=(ax)+1

以上是关于CMP分析流程的主要内容,如果未能解决你的问题,请参考以下文章