ATAC-seq专题---生信分析流程
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了ATAC-seq专题---生信分析流程相关的知识,希望对你有一定的参考价值。
参考技术A ATAC-seq信息分析流程主要分为以下几个部分:数据质控、序列比对、峰检测、motif分析、峰注释、富集分析,下面将对各部分内容进行展开讲解。下机数据经过过滤去除接头含量过高或低质量的reads,得到clean reads用于后续分析。常见的trim软件有Trimmomatic、Skewer、fastp等。fastp是一款比较新的软件,使用时可以用--adapter_sequence/--adapter_sequence_r2参数传入接头序列,也可以不填这两个参数,软件会自动识别接头并进行剪切。如:
fastp \
--in1 A1_1.fq.gz \ # read1原始fq文件
--out1 A1_clean_1.fq.gz \ # read1过滤后输出的fq文件
--in2 A1_2.fq.gz \ # read2原始fq文件
--out2 A1_clean_2.fq.gz \ # read2过滤后输出的fq文件
--cut_tail \ #从3’端向5’端滑窗,如果窗口内碱基的平均质量值小于设定阈值,则剪切
--cut_tail_window_size=1 \ #窗口大小
--cut_tail_mean_quality=30 \ #cut_tail参数对应的平均质量阈值
--average_qual=30 \ #如果一条read的碱基平均质量值小于该值即会被舍弃
--length_required=20 \ #经过剪切后的reads长度如果小于该值会被舍弃
fastp软件的详细使用方法可参考:https://github.com/OpenGene/fastp。fastp软件对于trim结果会生成网页版的报告,可参考官网示例http://opengene.org/fastp/fastp.html和http://opengene.org/fastp/fastp.json,也可以用FastQC软件对trim前后的数据质量进行评估,FastQC软件会对单端的数据给出结果,如果是PE测序需要分别运行两次来评估read1和read2的数据质量。
如:
fastqc A1_1.fq.gz
fastqc A1_2.fq.gz
FastQC会对reads从碱基质量、接头含量、N含量、高重复序列等多个方面对reads质量进行评估,生成详细的网页版报告,可参考官网示例:http://www.bioinformatics.babraham.ac.uk/projects/fastqc/good_sequence_short_fastqc.html
经过trim得到的reads可以使用BWA、bowtie2等软件进行比对。首先需要确定参考基因组fa文件,对fa文件建立索引。不同的软件有各自建立索引的命令,BWA软件可以参考如下方式建立索引:
bwa index genome.fa
建立好索引后即可开始比对,ATAC-seq推荐使用mem算法,输出文件经samtools排序输出bam:
bwa mem genome.fa A1_clean_1.fq.gz A1_clean_2.fq.gz
| samtools sort -O bam -T A1 > A1.bam
值得注意的是,在实验过程中质体并不能完全去除,因此会有部分reads比对到质体序列上,需要去除比对到质体上的序列,去除质体序列可以通过samtools提取,具体方法如下:首先将不含质体的染色体名称写到一个chrlist文件中,一条染色体的名称写成一行,然后执行如下命令即可得到去除质体的bam
samtools view -b A1.bam $chrlist > A1.del_MT_PT.bam
用于后续分析的reads需要时唯一比对且去重复的,bwa比对结果可以通过MAPQ值来提取唯一比对reads,可以用picard、sambamba等软件去除dup,最终得到唯一比对且去重复的bam文件。
比对后得到的bam文件可以转化为bigWig(bw)格式,通过可视化软件进行展示。deeptools软件可以实现bw格式转化和可视化展示。首先需要在linux环境中安装deeptools软件,可以用以下命令实现bam向bw格式的转换:
bamCoverage -b A1.bam -o A1.bw
此外,可以使用deeptools软件展示reads在特定区域的分布,如:
computeMatrix reference-point \ # reference-pioint表示计算一个参照点附近的reads分布,与之相对的是scale-regions,计算一个区域附近的reads分布
--referencePoint TSS \#以输入的bed文件的起始位置作为参照点
-S A1.bw \ #可以是一个或多个bw文件
-R gene.bed \ #基因组位置文件
-b 3000 \ #计算边界为参考点上游3000bp
-a 3000 \ #计算边界为参考点下游3000bp,与-b合起来就是绘制参考点上下游3000bp以内的reads分布
-o A1.matrix.mat.gz \ #输出作图数据名称
#图形绘制
plotHeatmap \
-m new_A1.matrix.mat.gz \ #上一步生成的作图数据
-out A1.pdf \ # 输出图片名称
绘图结果展示:
MACS2能够检测DNA片断的富集区域,是ATAC-seq数据call peak的主流软件。峰检出的原理如下:首先将所有的reads都向3'方向延伸插入片段长度,然后将基因组进行滑窗,计算该窗口的dynamic λ,λ的计算公式为:λlocal = λBG(λBG是指背景区域上的reads数目),然后利用泊松分布模型的公式计算该窗口的显著性P值,最后对每一个窗口的显著性P值进行FDR校正。默认校正后的P值(即qvalue)小于或者等于0.05的区域为peak区域。需要现在linux环境中安装macs2软件,然后执行以下命令:
macs2 callpeak \
-t A1.uni.dedup.bam \ #bam文件
-n A1 \ # 输出文件前缀名
--shift -100 \ #extsize的一半乘以-1
--extsize 200 \ #一般是核小体大小
--call-summits #检测峰顶信息
注:以上参数参考文献(Jie Wang,et.al.2018.“ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration.”Nature Communications)
ATAC分析得到的peak是染色质上的开放区域,这些染色质开放区域常常预示着转录因子的结合,因此对peak区域进行motif分析很有意义。常见的motif分析软件有homer和MEME。以homer软件为例,首先在linux环境中安装homer,然后用以下命令进行motif分析:
findMotifsGenome.pl \
A1_peaks.bed \ #用于进行motif分析的bed文件
genome.fa \ #参考基因组fa文件
A1 \ #输出文件前缀
-size given \ #使用给定的bed区域位置进行分析,如果填-size -100,50则是用给定bed中间位置的上游100bp到下游50bp的区域进行分析
homer分析motif的原理及结果参见:http://homer.ucsd.edu/homer/motif/index.html
根据motif与已知转录因子的富集情况可以绘制气泡图,从而可以看到样本与已知转录因子的富集显著性。
差异peak代表着比较组合染色质开放性有差异的位点,ChIP-seq和ATAC-seq都可以用DiffBind进行差异分析。DiffBind通过可以通过bam文件和peak的bed文件计算出peak区域标准化的readcount,可以选择edgeR、DESeq2等模型进行差异分析。
在科研分析中我们往往需要将peak区域与基因联系起来,也就是通过对peak进行注释找到peak相关基因。常见的peak注释软件有ChIPseeker、homer、PeakAnnotator等。以ChIPseeker为例,需要在R中安装ChIPseeker包和GenomicFeatures包,然后就可以进行分析了。
library(ChIPseeker)
library(GenomicFeatures)
txdb<- makeTxDbFromGFF(‘gene.gtf’)#生成txdb对象,如果研究物种没有已知的TxDb,可以用GenomicFeatures中的函数生成
peakfile <-readPeakFile(‘A1_peaks.narrowPeak’)#导入需要注释的peak文件
peakAnno <- annotatePeak(peakfile,tssRegion=c(-2000, 2000), TxDb=txdb)
# 用peak文件和txdb进行peak注释,这里可以通过tssRegion定义TSS区域的区间
对于peak注释的结果,也可以进行可视化展示,如:
p <- plotAnnoPie(peakAnno)
通过注释得到的peak相关基因可以使用goseq、topGO等R包进行GO富集分析,用kobas进行kegg富集分析,也可以使用DAVID在线工具来完成富集分析。可以通过挑选感兴趣的GO term或pathway进一步筛选候选基因。
JVM技术专题「原理专题」全流程分析Java对象的创建过程及内存布局
前言概要
对应过程则是:对象创建、对象内存布局、对象访问定位的三个过程。

对象的创建过程
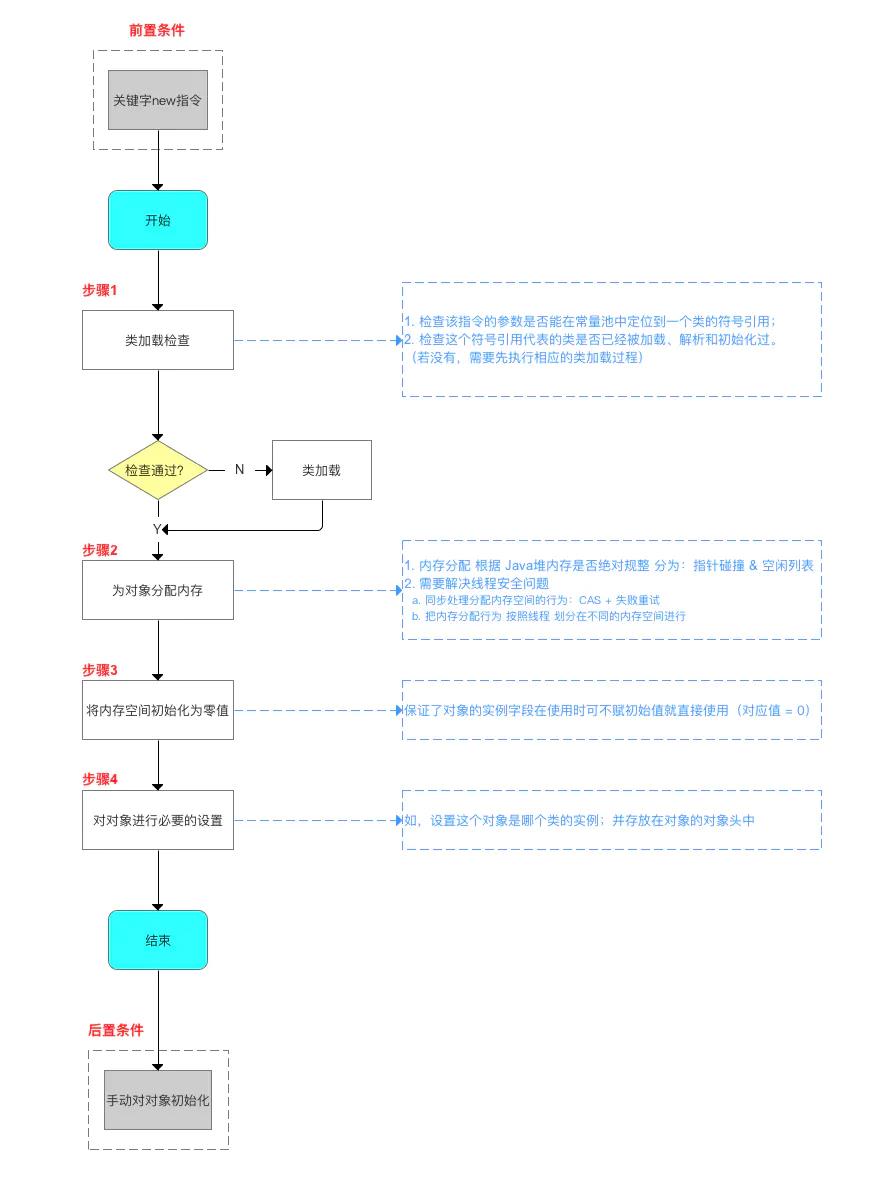
对象的创建方式
java中对象的创建方式有很多种,常见的是通过new关键字和反射这两种方式来创建。除此之外,还有clone、反序列化等方式创建。
通过new关键字创建
// Person zhangsan = new Person(id, height, weight)
Person zhangsan = new Person();
通过反射创建
反射创建对象,可以通过class.newInstance()调用无参的构造器创建对象,也可以使用构造器来创建constructor.newInstance()。
//Class clz = Class.forName("Person类的全限定类名")
Class clz = Person.class;
Person zhangsan = clz.newInstance()
// 使用构造器创建
Constructor<Person> cons = clz.getConstructor()
// 也可以指定参数类型获取有参构造器
Person zhangsan1 = cons.newInstance()
通过clone创建对象
当类实现了Cloneable接口时,可以使用clone()方法复制一个对象。需要留意是clone方法是浅拷贝。
Person libo = new Person(name: "李博", age:12, ...)
Person Livonor = new Person(name: "Livorno", age:32, ...)
libo.setFather(Livonor)
Person zhangsi = libo.clone() // 此时,张四和张三的名字、父亲在内存中都引用了相同的对象
反序列化创建
通过读取IO数据流创建,非本节重点
对象的创建过程
检查类加载(包含是否初始化、是否被加载、是否被解析)
对于new和反射两种创建方式而言,需要检查创建对象所使用的参数是否已完成类加载(比如它的类型和参数类型)。如果没有,要先完成类加载过程。
分配内存空间
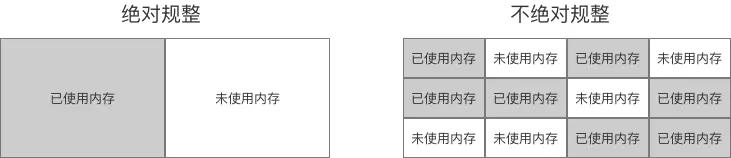
-
虚拟机为对象分配内存,即起始地址和偏移量。
-
对象所需要的空间在创建前就可以确定,但是起始地址需要在分配时去内存中找到一块足够大的空间。地址的分配有两种方式:指针碰撞和空闲列表。
指针碰撞

指针碰撞的方式是假设内存空间是规整的,被使用的和空闲的内存被分割成了两整块,通过一个指针记录分界点。在给对象分配内存的时候,将指针空闲区域移动一段与对象大小相等的距离即可。
空闲列表
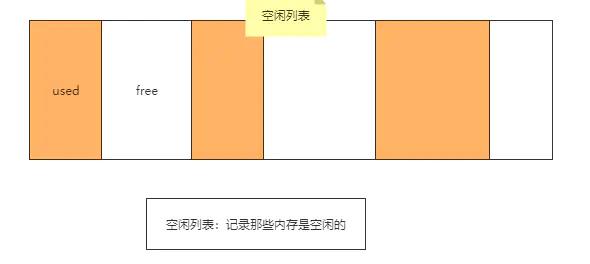
如果内存不规整,那么就需要维护一张表,来记录内存中那些地址是空闲的。分配对象时,通过空闲列表去找到一块足够大的空闲内存分配给对象并更新空闲列表。
多个线程创建的对象内存的冲突
举个例子,线程1和2同时要创建两个对象,指针是同一个。它们各自将指针加载到了cpu缓存,然后去执行分配地址空间的指令。结果就导致,后分配的哪一个,可能将先分配的那个对象的地址给覆盖了。
解决的办法有两种,一种是对分配内存的动作进行同步处理,即采用CAS加失败重试的方式,保证更新操作的原子性。
// 伪代码表示CAS+失败重试
while(true)
oldPtr = ptr //读取共享指针
newPtr = oldPtr + sizeOfInstance
if(compareAndSet(oldPtr, ptr, newPtr))break
另一种是使用TLAB的方式将线程的分配空间在堆内存中隔离开,在堆中为每个线程预先分配一小块不同的空间,每个线程创建对象都在自己对应的空间中完成。
即每个线程在 Java堆中预先分配一小块内存(本地线程分配缓冲(Thread Local Allocation Buffer ,TLAB)),哪个线程要分配内存,就在哪个线程的TLAB上分配,只有TLAB用完并分配新的TLAB时才需要同步锁。虚拟机是否使用TLAB,可以通过-XX:+/-UseTLAB参数来设定。
分配完内存之后,对象就已经存在于虚拟机的堆中了,此时虚拟机要将分配的内存空间初始化为零值(对象头例外)。
设置对象头
对象头包含了两种信息:MarkWord和类型指针。
-
MarkWord:存放对象本身的运行时状态数据(如HashCode, GC分代年龄、锁状态、是否偏向信息等)
-
KlassPointer:类型指针指向它的类型的元数据。对象头在对象的内存布局中细讲。
- 即对象指向它的类元数据的指针
- 虚拟机通过这个指针来确定这个对象是哪个类的实例
-
数据长度:如果对象 是 数组,那么在对象头中还必须有一块用于记录数组长度的数据
- 因为虚拟机可以通过普通Java对象的元数据信息确定对象的大小,但是从数组的元数据中却无法确定数组的大小。
执行对象实例构造函数
首先递归的执行父类的构造函数,然后收集本类中为实例变量赋值的语句并执行,最后执行构造方法中的语句。
public class AddA
public static void main(String[] args)
Father guy = new Son(30);
guy.saySomething();
System.out.println(guy.age);
class Father
int age = 60;
public Father()
saySomething();
public Father(int age)
this.age = age;
public void saySomething()
System.out.println("I am the father, " + age + "years old");
class Son extends Father
int age = 20;
public Son(int age)
// super(); 不写则隐式调用方法,写则必须在子类构造方法的第一句
saySomething();
this.age = age;
saySomething();
public void saySomething()
System.out.println("I am the son, " + age + " years old");
因为它涉及到了多态与方法的动态分派。在这里先简单描述一下它的执行过程,用来掌握构造方法的执行还是ok的。
-
首先,创建一个Son对象,然后调用其有参构造方法Son(int age)。
-
在有参构造方法中隐式调用了父类的无参构造方法,然后父类的构造方法继续调Object的构造方法。接下来收集为父类成员变量赋值的语句并执行。
-
由于多态中子类的成员变量会覆盖父类的成员变量,因此子类对象的age仍然是0。
-
同时无参构造方法中的saySomething()此时是被子类对象调用的,因此打印了第一句I am the son, 0 years old。
-
-
然后,super()方法出栈,回到子类构造方法中。此时应该收集为子类成员变量赋值的语句并执行。对象的age=20,saySomething()打印出第二句I am the son, 20 years old。
-
然后执行构造方法中的赋值语句int age = age;saySomething();
-
第三句话被打印I am the son, 30 years old。
-
-
子类对象创建完成,回到main方法。此时使用多态,将对象转成Father对象。
由于多态的规则:被重写的方法使用动态分派,查找(vtable)方法表,该方法实际是属于子类对象的。
因此guy.saySomething()实际调用的是子类对象的方法,打印出第四句话,I am the son, 30 years old。
最后,输出guy.age. 成员变量不具备多态性,因此打印出父类对象的age 60.
I am the son, 0 years old
I am the son, 20 years old
I am the son, 30 years old
I am the son, 30 years old
对象的内存布局

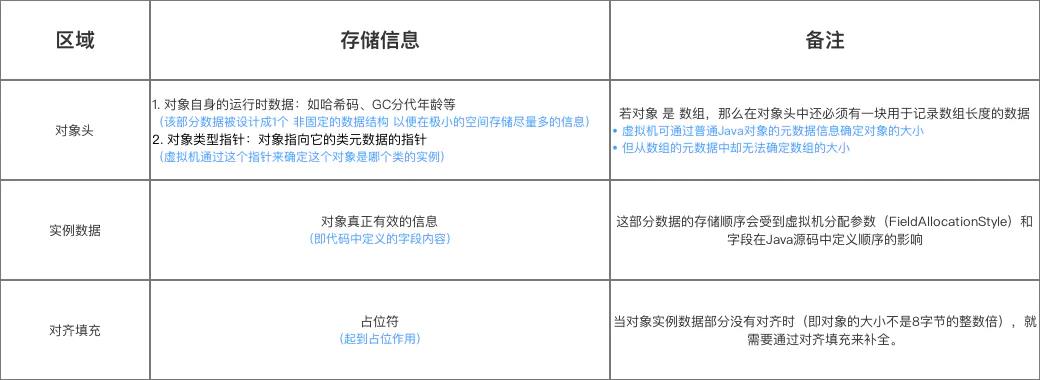
对象在堆中的存储布局划分为三个部分:对象头、实例数据和对其填充(padding)。
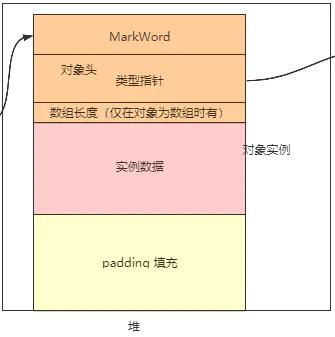
对象的内存布局
对象头
对象头中包含markword(标记字段)和类型指针【数组长度】。
markword
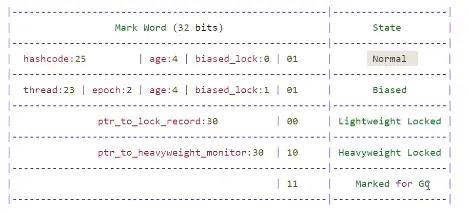
markword存储与对象自身定义数据无关的信息,用来表示对象的运行时状态。包括了HashCode,GC年龄,锁状态等信息。在一个32位的虚拟机中,markword用一个32位的bitmap表示,bitmap最后两位存放锁状态信息,如下图。
-
markword
-
普通状态下,状态为01,存储hashcode,分代年龄,偏向锁状态为0。
-
偏向锁状态下,状态为01,存储持有偏向锁的线程和重入次数,分代年龄,偏向锁状态为1。此时hashcode没了,但是,hashcode可以通过Object的hashcode()方法计算出来,只要没有重写该方法,那么得到的哈希码始终是一致的。
-
轻量级锁,状态为00。通过cas方式将对象的markword信息原子性地交换到了持有该对象锁的线程中,存储在lockRecord内,并同时将lockRecord的指针存放在对象头Markword的前30位。
-
重量级锁状态下,前30位存放指向锁控制器Monitor的指针,锁状态为10.
-
对象被标记为待回收状态时,最后两位状态为11.
-
-
KlassPointer(类型指针)
指向类型元数据,从而可以通过对象来访问到它的类型信息。
- 数组长度(array length)
主要记录数组的长度信息一般为4字节(根据int的范围来考虑)
实例数据
-
实例数据中存放了对象的字段信息。无论是从父类继承的,还是在子类中定义的,都保存在实例数据中。
-
按照一定顺序存放,在满足这个顺序的条件下,父类定义的字段又会出现在子类定义的变量之前。
即代码中定义的字段内容
注:这部分数据的存储顺序会受到虚拟机分配参数(FieldAllocationStyle)和字段在Java源码中定义顺序的影响。
// HotSpot虚拟机默认的分配策略如下:
longs/doubles、ints、shorts/chars、bytes/booleans、oop(Ordinary Object Pointers)
- // 从分配策略中可以看出,相同宽度的字段总是被分配到一起
- // 在满足这个前提的条件下,父类中定义的变量会出现在子类之前
CompactFields = true;
// 如果 CompactFields 参数值为true,那么子类之中较窄的变量也可能会插入到父类变量的空隙之中。
对齐填充
如果对象的实例数据占用空间不是8的整数倍,则填充0值让对象的占用空间位8的整数倍。
对象的访问定位
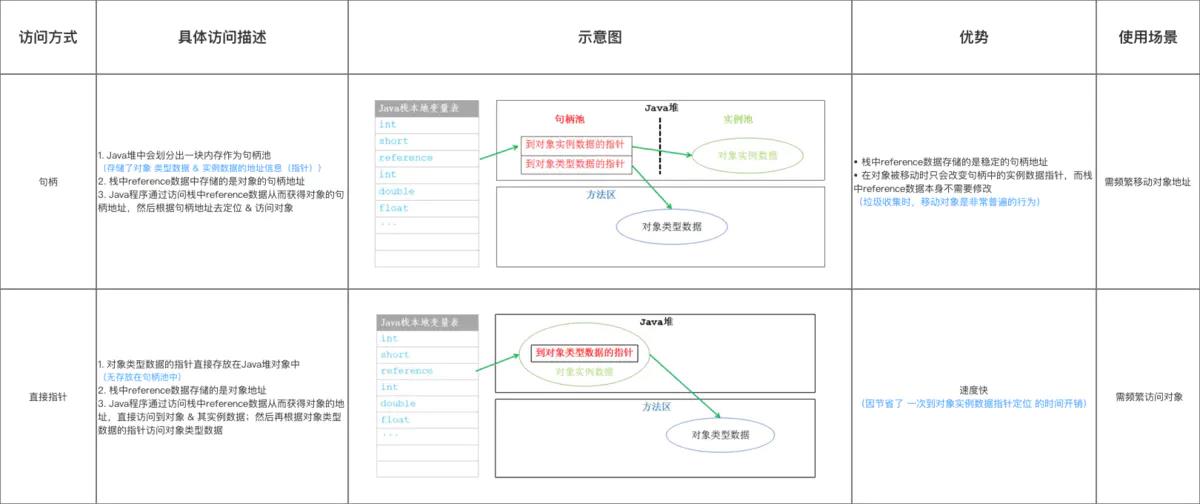
常见的有两种方式,句柄访问和直接指针访问。
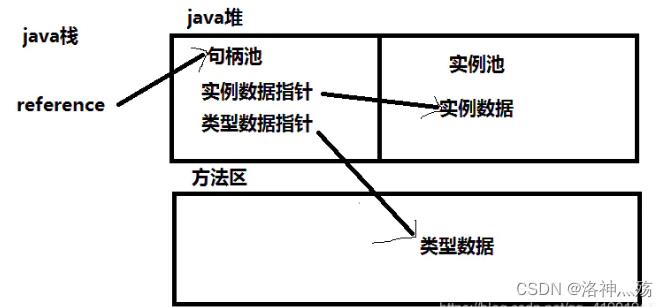
句柄访问
-
使用句柄访问的话,对象的引用(如zhangsan),指向的是句柄池中的某个句柄,该句柄存放了指向实际实例对象的指针和指向方法区数据类型的指针。
- 其好处是,当对象被移动的时候(比如垃圾回收时,整理内存空间需要大量移动对象),不需要频繁的修改引用,只需要修改句柄中实例数据指针。
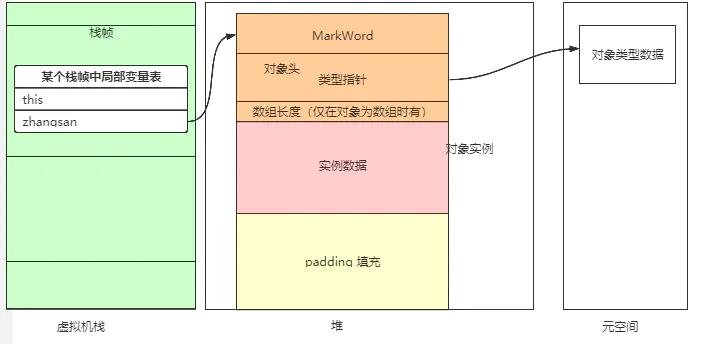
- 通过指针访问,则是对象的引用直接指向了该对象。其好处是,通过引用访问对象时,不需要多一次的指针定位,使得访问速度更快。
以上是关于ATAC-seq专题---生信分析流程的主要内容,如果未能解决你的问题,请参考以下文章