一个核糖体与mRNA的结合部位,为啥形成2个tRNA结合位点
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了一个核糖体与mRNA的结合部位,为啥形成2个tRNA结合位点相关的知识,希望对你有一定的参考价值。
参考技术A 在翻译过程中,核糖体有多个结合位点以及催化位点,保证翻译能顺利、快速的进行,核糖体与trna的结合位点有3个,并不能说核糖体与mrna的结合位点也相应的有3个,但因两者有着紧密的联系,因此,人教版高中生物教材为了降低学生的学习难度,削减了许多繁琐的内容,简化教学知识,就采用了“核糖体与mrna的结合部位会形成2个trna结合位点”这样的语言来进行描述。具体原因如下:
肽链的延伸循环可分为三个阶段:进位、肽键形成和移位。进位是指aa-trna(氨酰trna)进入a位。aa-trna是以eef-1a·gtp·aa-trna复合物的形式进人a位的,并需要gtp的水解。在这一过程中,如何保证正确aa-trna的进入即翻译的准确性是一关键问题。目前有两种模型,一种认为对aa-trna经过了二步选择,第一步是在三元复合物进入a位时;第二步是在gtp水解后和肽键形成前。另一种是三位点模型,认为除a和p位外,还存在一个aa-trna结合位点。在原核生物三个位点(e位点)已被证实,而真核生物资料还不充分。已发现兔肝80s核糖体有三个aa-trna结合位点。
核糖体有3个trna的结合位点,分别称为a、p和e位点。
蛋白质合成开始时,先由一个较小的核糖体亚基(30s)结合到mrna,在一些起始蛋白质——
σ
因子等的协助下,构成一个30s—mrna起始复合体。
真核生物mrna具有m7gpppnp帽子结构,核糖体上有专一位点或因子识别mrna的帽子,使mrna与核糖体结合。
在多肽合成过程中,不同的trna将相应的氨基酸带到蛋白质合成部位,并与mrna进行专一性的相互作用,以选择对信息专一的aa-trna(氨酰trna)。核糖体还必须能同时容纳另一种携带肽链的trna,即肽基mtrna(peptidyl-trna),并使之处于肽键易于生成的位置上。核糖体必须包括至少5个活性中心,即mrna结合部位、结合或接受aa-trna部位(a位)、结合或接受肽基trna的部位、肽基转移部位(p位)及形成肽键的部位(转肽酶中心)。此外,还应有负责肽链延伸的各种延伸因子的结合位点。核糖体小亚基负责对模板mrna进行序列特异性识别,如起始部分识别、密码子与反密码子的相互作用等,mrna的结合位点也在小亚基上。大亚基具有携带氨基酸和trna的功能。此外,肽键的形成、aa-trna、肽基-trna的结合位、p位、转肽酶中心等都在大亚基上完成。
从以上资料可知,在翻译过程中,核糖体有多个结合位点以及催化位点,保证翻译能顺利、快速的进行,核糖体与trna的结合位点有3个,并不能说核糖体与mrna的结合位点也相应的有3个,但因两者有着紧密的联系,因此,人教版高中生物教材为了降低学生的学习难度,削减了许多繁琐的内容,简化教学知识,就采用了“核糖体与mrna的结合部位会形成2个trna结合位点”这样的语言来进行描述。
[生物信息学]分子生物学的核心法则
[生物信息学]分子生物学的核心法则
主要用 python 实现分子生物学中核心法则的基础功能,入门第一课。当然,核心部分还是计算,所以会轻放生物部分。
这里学习的核心法则指的是 蛋白质生物合成,即 脱氧核糖核酸(DNA) 通过 转录(transcribe) 获得 信使核糖核酸(mRNA),mRNA 再通过 转译(translate) 获得 蛋白质(protein) 的过程。
过程图解如下:
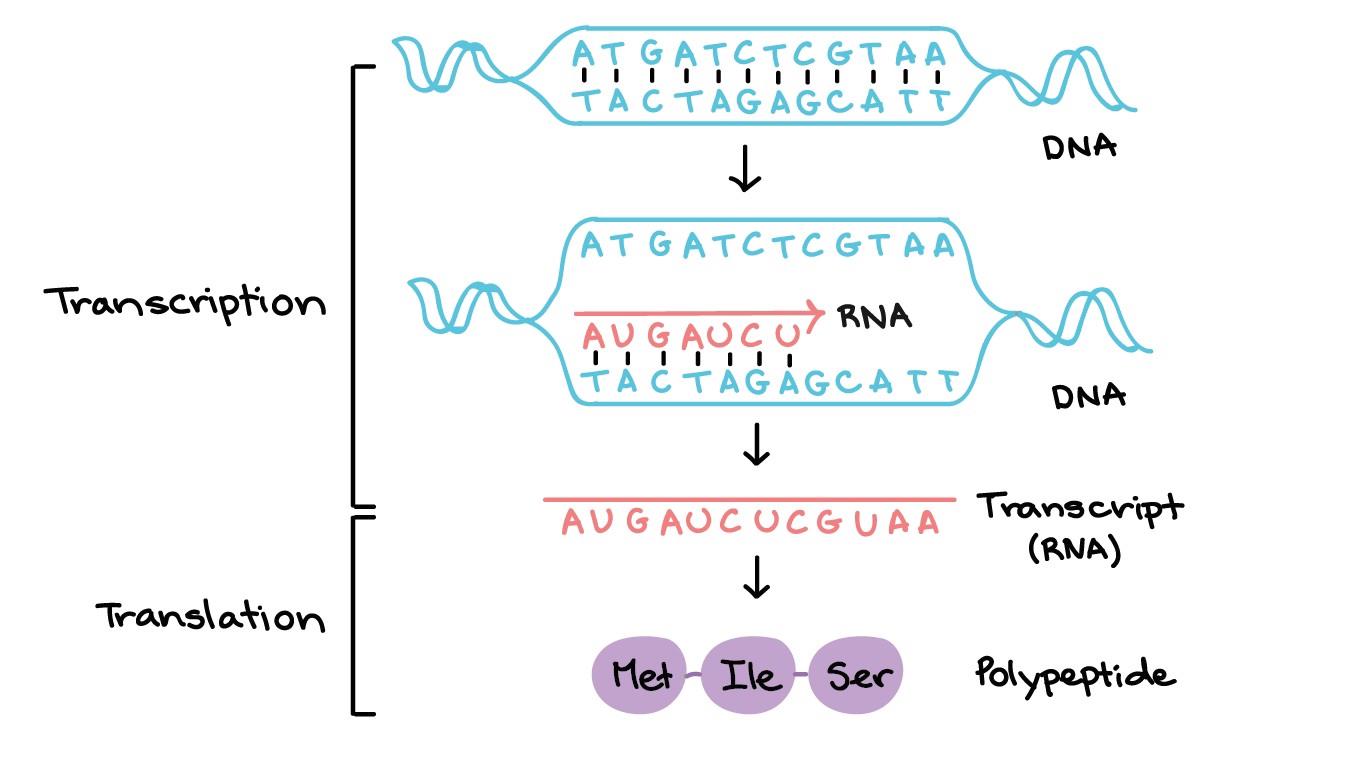
DNA 的反向互补(reverse complement)
DNA 由四种碱基组成:A, G, T, C,其中 A 与 T 互补,G 与 C 互补。而 DNA 又是由两条 DNA 链由中心轴盘绕,形成双螺旋结构,两条 DNA 链反向互补,形成稳定的结构:

图像资料来源于: https://simple.wikipedia.org/wiki/Complementarity_(molecular_biology)
计算方式为:
-
获取一条 DNA 链的补数
如
ATCG,它的补数为TAGC。 -
进行反转
TAGC反转为CGAT
python 部分:
dna_pair = {
'A': 'T',
'T': 'A',
'C': 'G',
'G': 'C'
}
def reverse_complement(sequence):
return ''.join([dna_pair[gene] for gene in sequence])[::-1]
本质上来说这一段的写法是这个意思:
def reverse_complement(sequence):
# 这里新建一个新的数组去保留所有的值
reversed_sequence = []
# 遍历所有的序列,找到对应的碱基
for gene in sequence:
reversed_sequence += dna_pair[gene]
# [::-1] 起到翻转 字符串/数组 的作用
# ''.join() 起到将数组转成字符串的作用
return ''.join(reversed_sequence)[::-1]
转录(transcribe)
转录是 DNA 变为 mRNA 的过程,与 DNA 的反向互补 过程相似,只不过 A 与 U 互补,而且不需要翻转。
python 部分:
rna_pair = {
'A': 'U',
'T': 'A',
'C': 'G',
'G': 'C'
}
def transcribe(sequence):
return ''.join([rna_pair[gene] for gene in sequence])
最后的结果是 ATCG 会转为 UAGC。
转译(translate)
即 mRNA 转为 蛋白质 的过程。
mRNA 三个基因为一组转换为对应的氨基酸,其配对表如下:
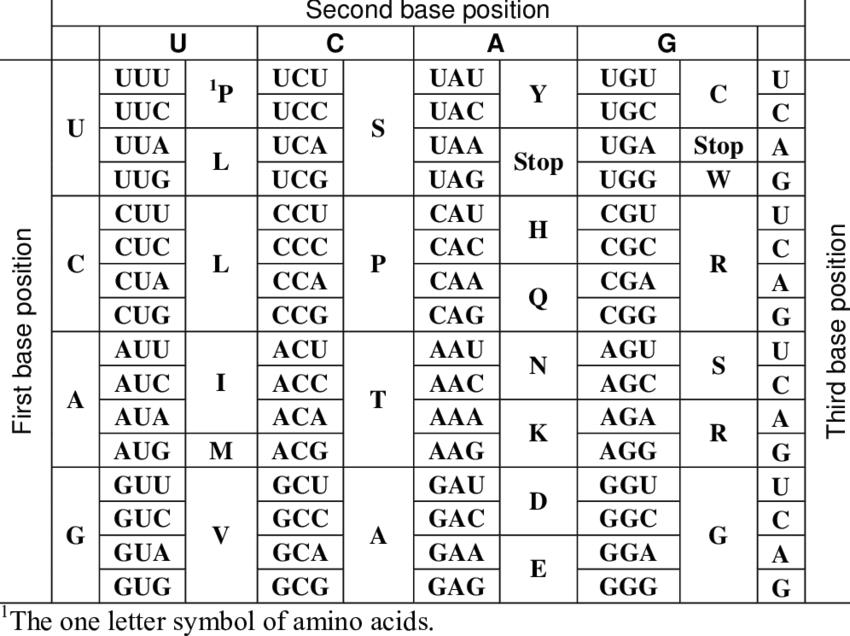
图像资料来源于: https://www.researchgate.net/figure/The-standard-genetic-code-table_tbl1_7002668
python 实现如下:
# 首先有 mRNA 与 氨基酸的对应表
# https://www.geeksforgeeks.org/dna-protein-python-3/
genetic_code = {
'ATA':'I', 'ATC':'I', 'ATT':'I', 'ATG':'M',
'ACA':'T', 'ACC':'T', 'ACG':'T', 'ACT':'T',
'AAC':'N', 'AAT':'N', 'AAA':'K', 'AAG':'K',
'AGC':'S', 'AGT':'S', 'AGA':'R', 'AGG':'R',
'CTA':'L', 'CTC':'L', 'CTG':'L', 'CTT':'L',
'CCA':'P', 'CCC':'P', 'CCG':'P', 'CCT':'P',
'CAC':'H', 'CAT':'H', 'CAA':'Q', 'CAG':'Q',
'CGA':'R', 'CGC':'R', 'CGG':'R', 'CGT':'R',
'GTA':'V', 'GTC':'V', 'GTG':'V', 'GTT':'V',
'GCA':'A', 'GCC':'A', 'GCG':'A', 'GCT':'A',
'GAC':'D', 'GAT':'D', 'GAA':'E', 'GAG':'E',
'GGA':'G', 'GGC':'G', 'GGG':'G', 'GGT':'G',
'TCA':'S', 'TCC':'S', 'TCG':'S', 'TCT':'S',
'TTC':'F', 'TTT':'F', 'TTA':'L', 'TTG':'L',
'TAC':'Y', 'TAT':'Y', 'TAA':'_', 'TAG':'_',
'TGC':'C', 'TGT':'C', 'TGA':'_', 'TGG':'W',
}
# 随后进行转换
def translate_rna_fragment(rna_sequence):
decoded_str = []
sequence_list = list(rna_sequence)
while (len(sequence_list) >= 3):
decoded_str += genetic_code[''.join(sequence_list[:3])]
sequence_list = sequence_list[3:]
return ''.join(decoded_str)
注*:不直接用字符串而是转为数组的原因在于,python 中字符串是不可变的,所以每一次更新字符串的代价都很大。作业体量小的时候问题不大,一旦作业体量比较大的话——如果是真的生物,最小的基因组也是在百万起跳,运行速度肯定不尽如人意。
本章学习内容主要重点还在 字符串/数组 的操作,以及 字典 的取值,算是对 python 的一个入门级学习。
以上是关于一个核糖体与mRNA的结合部位,为啥形成2个tRNA结合位点的主要内容,如果未能解决你的问题,请参考以下文章