GWAS研究中样本数量和结果真实有效性之间是怎样的?
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GWAS研究中样本数量和结果真实有效性之间是怎样的?相关的知识,希望对你有一定的参考价值。
GWAS原理部分可以参考Fisher\'s exact test,把链接里的例子中,男人/女人替换成有病/没病,节食替换成SNP/CNV,给出的p值表示SNP与疾病如果是完全独立的,那么由随机分布导致的odd ratio比现在更显著的概率。1e-8的p值可能看起来很显著,但考虑到人类的SNP数量可能在千万级,综合Multiple comparisons problem的校正的话,那么这个p值一点都不显著。这就是一个由数据得出结论的纯统计问题,说不上什么硬伤,花精力啃下多重检验校正和假阳性率的统计资料。人类的GWAS热潮已过了,要说有什么效果的话,就是砸了钱打了不少水漂,得到的结果相对投入来讲,寥寥。农业上GWAS现在很热,原因一方面取样比人简单,性状也容易标准化,且大多只当作一个育种筛选的分子marker,很少再继续研究功能的。要做SNP的话,技术有很多,主要划分是考虑通量与成本。测序做SNP主要是resequencing做新SNP发现这块。二代是主要手段,因为成本。三代的优势在于,当需要考虑phasing信息,即数kb片段内的SNP位点连锁信息时,是唯一的选择。resequencing是resequencing,de novo是de novo,这是两个问题,de novo一律都是选片段长的效果更好,当然考虑成本的话,大家都懂。做复杂的结构变异的话,在不同的尺度上有不同的技术,二代理论上都可以做,但实际上在每个特定尺度下,都不是最好的选择。做拷贝数变异的话,那么最好的技术,是OncoScanHD(芯片)上使用的倒置探针技术,线性范围可以到50倍。简单的串联重复到数kb内的变异,三代优势项目。数十kb到兆b级的,最佳技术是Bionano Irys光学图谱系统,再大的话,手段就更多了。单倍型解析主要依赖的是高质量的SNP数据库,程序算法之类的话,考虑性能和通量其实也就那几个可以选。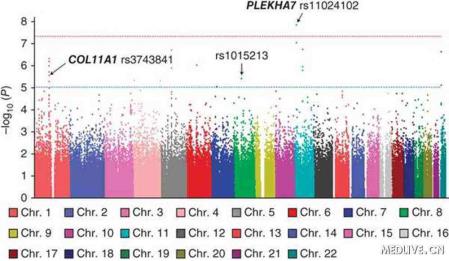
“取到足够低的P值”本身不是目的,目的是“检测到统计上可信的效应”。我觉得这部分属于Power Analysis的范畴:检测一个效应时,为了达到一定的可信度(比如说用P值来度量可信度),如果随机干扰越大,效应越小,需要的样本量就越大。需要多大的样本是可以通过公式估算的。GWAS研究中随机干扰往往很大。可能的干扰包括遗传背景的不同,环境效应的不同等等。如果题主所说的疾病/体质有人研究过遗传力/遗传参数,可以对这个随机干扰的大小有个初步的估计。假如世界上只有两个人名字叫做“就我们俩”,然后全世界只有他们两个得了某种叫做“很少见的病”的病,楼主敢相信这种病是由于这个名字造成的吗?这里讨论的并不是“名字究竟会不会致病”,而是“一个统计上极高的关联”究竟能够代表什么。
GWAS基因芯片数据预处理:质量控制(quality control)
一、数据为什么要做质量控制
比起表观学研究,GWAS研究很少有引起偏差的来源,一般来说,一个人的基因型终其一生几乎不会改变的,因此很少存在同时影响表型又影响基因型的变异。但即便这样,我们在做GWAS时也要去除一些可能引起偏差的因素。
这种因素主要有:群体结构、个体间存在血缘关系、技术性操作。
二、怎么看数据是否需要进行质量控制
下面分别为样本和SNP位点在数据中的直方图,当数据不在绝大多数的分布当中时,我们会倾向于认为那是测序、人工操作等其他方面造成的误差,而非该个体的真实情况,因此是需要将这些样本和位点过滤掉的。
这个阈值的设定并没有一个金标准,可参考往年发表的文献的常用阈值。
1、样本过滤阈值的设定
2、SNP过滤阈值的设定
三、怎么进行质量控制
质量控制包括两个方向,一个是样本的质量控制,一个是SNP的质量控制
1、样本的质量控制
样本的质量控制包括:缺失率、杂合性、基因型性别和记录的性别是否一致。
1)检测缺失率,通常情况下,将样本缺失率大于5%的个体去除
plink --bfile file --mind 0.05 --make-bed --out file_mind
2)检测杂合性
plink --bfile file --het --make-bed --out file_het
3) 检测性别不一致的个体
plink --bfile file --check-sex --make-bed --out file_checksex
4)去除不符合的样本
将1-3)获得不符合的样本去除
plink --bfile removesample.txt --remove removesample.txt --make-bed --out file_qcsample
removesample.txt的格式如下:
FID IID
ASN ind1
ASN ind2
2、SNP位点的质量控制
SNP位点的质量控制包括:MAF值、call出率、Hardy-Weinberg Equilibrium
其命令见如下:
plink --bfile file_mind_file_qcsample --hwe 0.00001 --geno 0.02 --maf 0.01 --make-bed --out file_qcsample_snp
--hwe指的是不符合哈温伯格平衡的SNP位点,P值小于0.00001;
--geno指的是基因型缺失率大于2%的样本;
--maf指的是次等位基因频率低于1%的SNP位点;
最后,会得出干净的SNP和样本。
文中图片出处:
https://jvanderw.une.edu.au/Mod2Lecture_PLINK.pdf
以上是关于GWAS研究中样本数量和结果真实有效性之间是怎样的?的主要内容,如果未能解决你的问题,请参考以下文章