【java】-关于String的使用以及其输出结果的问题
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了【java】-关于String的使用以及其输出结果的问题相关的知识,希望对你有一定的参考价值。
RT... 输出的结果为什么会是这种形式? 求解
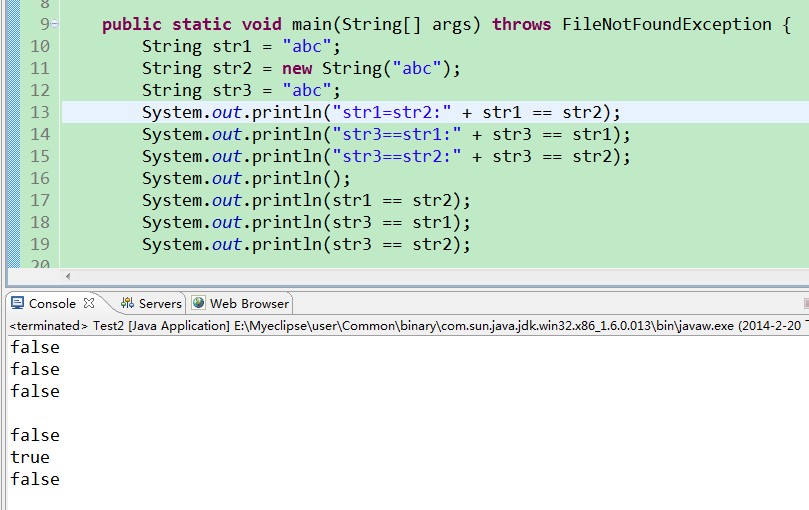
==是比较内存地址的
String的两种创建方式
String str1=“abc”;在字符串常量池中生成abc ,如果有不生成直接引用。
所以str1与str3地址一样
String str2=new String("abc");在堆中生成String对象其值为“abc”
如果用equals比较所有结果都为true
追问我只是想知道为什么前三句中的字符串不输出,并且第二句不是应该为true么,怎么也变在了false.
追答+号的作用啊 以“str1==str3”+str1==str3为例
先进行左结合 "str1==str3"+str1生成新的字符串str1==str3abc之后在于str3比较。
想要你的结果需要用括号"str1==str3"+(str1==str3)
首先,你要知道String类型在Java里属于引用传递类型,即不可变的;String不属于基本数据类型中的一种。
其次,是关于String的两种实例方式。String str = "abc";只会生成一个对象,如果之前有的话;String str = new String("abc");则会生成两个对象。
最后,回答你的疑问:在开始时,str1和str3比较前为false,比较后位true,是因为引用传递和创建对象时开辟内存地址造成的。 参考技术C 前面的字符串和后面的字符串连接形成新的字符串,再与最后的字符串进行比较呗 参考技术D 字符串比较要用
boolean
equals(Object anObject)
将此字符串与指定的对象比较。
bwa比对软件的使用以及其结果文件(sam)格式说明
一、bwa比对软件的使用
1、对参考基因组构建索引
bwa index -a bwtsw hg19.fa # -a 参数:is[默认] or bwtsw,即bwa构建索引的两种算法,两种算法都是基于BWT的(BWT search while the CIGAR string by Smith-Waterman alignment.)。-a bwtsw对于短的参考序列是不工作的,必须要大于等于10Mb;-a is 不适用于大的参考序列,必须要小于等于2G;
output:hg19.fa.amb、hg19.fa.ann、hg19.fa.bwt、hg19.fa.pac和hg19.fa.sa
2、寻找输入reads文件的SA坐标
对于pair end数据,每个reads文件单独做运算,single end数据就不用说了,只有一个文件。
pair end:
bwa aln hg19.fa read1.fq.gz -l 30 -k 2 -t 4 -I > read1.fq.gz.sai or bwa aln hg19.fa read1.fq.gz -l 30 -k 2 -t 4 -I -f read1.fq.gz.sai
bwa aln hg19.fa read2.fq.gz -l 30 -k 2 -t 4 -I > read2.fq.gz.sai or bwa aln hg19.fa read2.fq.gz -l 30 -k 2 -t 4 -I -f read2.fq.gz.sai
single end:
bwa aln hg19.fa read.fq.gz -l 30 -k 2 -t 4 -I > read.fq.gz.sai or bwa aln hg19.fa read.fq.gz -l 30 -k 2 -t 4 -I -f read.fq.gz.sai
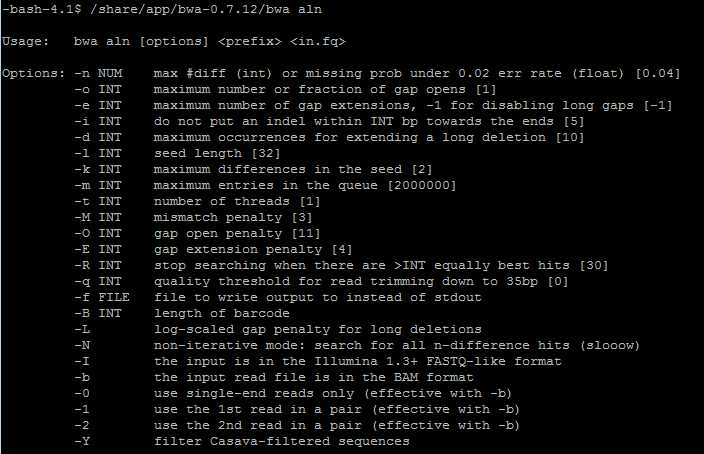
主要参数说明:
-o int:允许出现的最大gap数。
-e int:每个gap允许的最大长度。
-d int:不允许在3’端出现大于多少bp的deletion。
-i int:不允许在reads两端出现大于多少bp的indel。
-l int:Read前多少个碱基作为seed,如果设置的seed大于read长度,将无法继续,最好设置在25-35,与-k 2 配合使用。
-k int:在seed中的最大编辑距离,使用默认2,与-l配合使用。
-t int:要使用的线程数。
-R int:此参数只应用于pair end中,当没有出现大于此值的最佳比对结果时,将会降低标准再次进行比对。增加这个值可以提高配对比对的准确率,但是同时会消耗更长的时间,默认是32。
-I int:表示输入的文件格式为Illumina 1.3+数据格式。
-B int:设置标记序列。从5’端开始多少个碱基作为标记序列,当-B为正值时,在比对之前会将每个read的标记序列剪切,并将此标记序列表示在BC SAM 标签里,对于pair end数据,两端的标记序列会被连接。
-b :指定输入格式为bam格式。bwa aln hg19.fa read.bam > read.fq.gz.sai
3、生成sam格式的比对文件
如果一条read比对到多个位置,会随机选择一种
single end:bwa samse hg19.fa read.fq.gz.sai read.fq.gz > read.fq.gz.sam
参数:
-n int:如果reads比对次数超过多少次,就不在XA标签显示。
-r str:定义头文件。‘@RG\\tID:foo\\tSM:bar’,如果在此步骤不进行头文件定义,在GATK后续分析中还是需要重新增加头文件。
pair end:bwa sampe -a 500 read1.fq.gz.sai read2.fq.gz.sai read1.fq.gz read2.fq.gz > read.sam
参数:
-a int:最大插入片段大小。
-o int:pair end两reads中其中之一所允许配对的最大次数,超过该次数,将被视为single end。降低这个参数,可以加快运算速度,对于少于30bp的read,建议降低-o值。
-r str:定义头文件。同single end。
-n int:每对reads输出到结果中的最多比对数。
4、其他
(1)
bwa mem ref.fa reads.fq > aln-se.sam 单端测序
bwa mem ref.fa read1.fq read2.fq > aln-pe.sam 双端测序
(2)
bwa aln ref.fa short_read.fq > aln_sa.sai
bwa samse ref.fa aln_sa.sai short_read.fq > aln-se.sam
bwa sampe ref.fa aln_sa1.sai aln_sa2.sai read1.fq read2.fq > aln-pe.sam
(3)
bwa bwasw ref.fa long_read.fq > aln.sam
二、sam文件格式说明
1、
XT:A:U/R Type:Unique/Repeat/N/Mate-sw # U指第五列比对值>0;R指第五列比对值==0
参考文献:
1、《GATK使用方法详解(包含bwa使用)》http://www.tanboyu.com/gatk-bwa.html
2、《bwa英文操作手册》http://www.chinadmd.com/file/ecaeoaecwzvs3trpxpwtzows_1.html
以上是关于【java】-关于String的使用以及其输出结果的问题的主要内容,如果未能解决你的问题,请参考以下文章