mysql怎么把语言改成中文
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了mysql怎么把语言改成中文相关的知识,希望对你有一定的参考价值。
mysql把语言改成中文的步骤如下:
第一步我们需要打开软件,创建一个数据库,如下图所示:
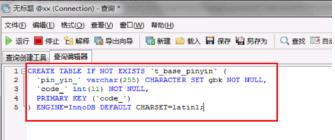
第二步创建数据库之后,需要创建一张汉字和拼音对照表,使用create table语句创表,如下图所示:
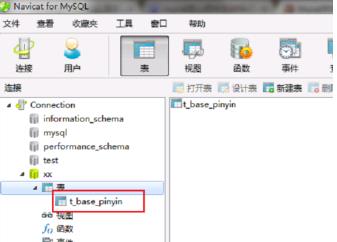
第三步我们打开表,可以看到成功创建一张名为t_base_pinyin的表,并且含有pin_yin_和code_两个字段,如下图所示:
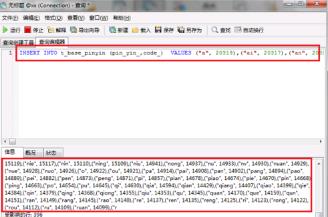
第四步我们使用INSERT INTO t_base_pinyin () VALUES ()语句来给汉字拼音对照表添加对照数据,如下图所示:
第五步我们打开t_base_pinyin ,可以看到对照数据已经插入成功,如下图所示:
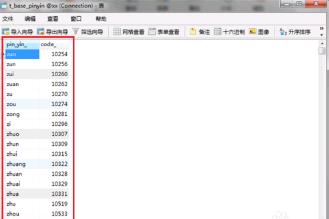
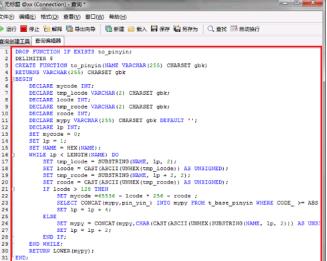
第六步我们最后需要创建一个拼音转换汉字的函数,输入完成之后进行执行,语句如下图所示:
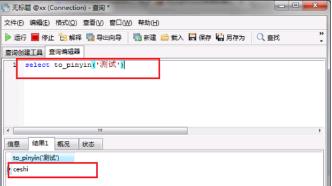
第七步我们输入select to_pinyin('测试')语句进行查询,可以看到输出to_pinyin('测试'),ceshi,已经成功将中文转换成拼音,如下图所示:
2、如果安装的时候未选,可以在创建数据库时指定编码,比如:
create database mydb character set utf8; 参考技术B 将mysql的编码格式改为utf-8,就可以了。具体修改步骤网上查本回答被提问者采纳
r语言孟德尔随机化分析,怎么把ivw改成随机效应模型
孟德尔随机化(Mendelian randomization,MR)是以孟德尔独立分配定律为基础进行流行病学研究设计和数据分析,论证病因假说的一种方法。由基因型决定中间表型(暴露)的差异, 因果方向明确。通过引入一个称之为工具变量的中间变量,来分析暴露因素和结局之间的因果关系
2.孟德尔随机化 vs RCT
孟德尔随机化的目的不是估计遗传效应的大小,而是估计暴露对结果的因果效应,所以与遗传变异相关的结局的平均变化幅度可能与干预措施导致的变化幅度不同
即使遗传变异与结果之间的关联程度很小,暴露的人群归因风险也不一定很低,因为暴露可能会比遗传变异解释更大的变化程度(例如,他汀类药物对低密度脂蛋白胆固醇水平的影响比低密度脂蛋白胆固醇水平与HMGCR基因变异的关联要大几倍,因此对后续结果的影响更大。)
孟德尔随机化要求大样本研究,变异发生率不能太小(最小等位基因频率MAF>5%)
3.工具变量
工具变量本身是一个计量经济学的概念,在孟德尔随机中,遗传变异被用作工具变量评估暴露对结局的因果效应,遗传变异满足工具变量的基本条件总结为(孟德尔随机化核心假设):
关联性假设——遗传变异与暴露有关
独立性假设——该遗传变异与暴露-结果关联的任何混杂因素均不相关
排他性假设——该遗传变异不会影响结果,除非可能通过与暴露的关联来实现
某研究组想了解非洲村落里的儿童补充维生素A和其死亡情况的关联,如果仅仅利用维生素A的服用情况和死亡情况去判断两者的关联,那极有可能会产生很大的偏倚,这是因为维生素A的服用情况和很多潜在因素相关,比如家庭的经济困难程度、家庭成员以及实验儿童的依从性,而这些潜在的因素也可能对儿童的身体健康有很大的影响。因此,在研究起始设计中,研究者便利用工具变量来解决这个问题。
在这里,工具变量Z是指服用维生素A这个任务,类似于随机抽签。这样的话工具变量Z便只和X服用维生素A这个行为相关,与除X以外的混杂因素不相关。
4.应用范围
行为因素与健康:基因变异引起各个倾向某行为,决定暴露状态。如ALDH2变异引起乙醛代谢障碍,改变饮酒行为,不同ALDH基因型代表饮酒量多少;
机体代谢产物与疾病关系,估计长期效应。代谢产物是基因表达的中间表型,酶的底物或者体外难测量的代谢指标:如LDL受体基因变异引起家族高胆固醇血症,比较不同基因型之间CHD发病情况的差异,可模拟血胆固醇水平和CHD发病关系;
子宫内环境暴露于子代健康关系。
5.发文分析
孟德尔随机化研究均发表在影响因子5分以上的期刊中
6.基础分析流程——TwoSampleMR
找工具变量,我们要的是基因作为工具变量,这些基因都是从别人的研究中挑出来的,所有的基因研究有个专门的库叫做genome wide association studies (GWAS)。我们需要做的就是从这个库中挑出来我们自己需要的和我们暴露相关的基因变量SNPs。
估计工具变量对结局的作用,工具变量对结局的作用也是从所有的研究中估计出来的整体效应,这样可以拒绝单个研究的偏倚。
合并多个SNP的效应量,这个效应量是我们得到暴露和结局因果效应的前提。
处理数据,用合并后的数据进行孟德尔随机化分析和相应的敏感性分析。
7.TwoSampleMR代码实现
安装相关R包
install.packages('devtools')
library('devtools')
install_github("MRCIEU/TwoSampleMR") #安装TwoSampleMR包
library('TwoSampleMR')
devtools::install_github("mrcieu/ieugwasr",force = TRUE)
获取MR base的表型ID,将结果保存为pheno_info.csv这个文件
ao <-available_outcomes(access_token=NULL) #获取GWAS数据,但近期Google限制,容易被墙
write.csv(ao,'pheno_info.csv',row.names=F)#将数据写入本地存储
查看pheno_info.csv文件,获取与暴露相关的工具变量的信息以及结局信息。这里选择暴露为obesity class 2 (ID = 91), 结局为 type 2 diabetes (ID = 1090)
exp_dat <- extract_instruments(outcomes=91,access_token=NULL)
obesity_exp_dat <- clump_data(exp_dat)
t2d_out_dat <- extract_outcome_data(snps=obesity_exp_dat$SNP, outcomes=1090, access_token=NULL)#提取结果信息
dat <- harmonise_data(exposure_dat =obesity_exp_dat, outcome_dat= t2d_out_dat)#数据合并,计算基因对结局的合并效应量
孟德尔随机化
results <- mr(dat)
OR值
OR <- generate_odds_ratios(results)
异质性检验
heterogeneity<- mr_heterogeneity(dat)
多效性检验
pleiotropy<- mr_pleiotropy_test(dat)
逐个剔除检验
leaveoneout<- mr_leaveoneout(dat)
散点图
mr_scatter_plot(results,dat)
森林图
results_single<- mr_singlesnp(dat)
mr_forest_plot(results_single)
漏斗图
mr_funnel_plot(results_single)
实例解析
2022年10月10日
西安交通大学生物医学信息与基因组学中心杨铁林教授团队在Nature Neuroscience (IF=28.771)期刊发表了题为:Mendelian randomization analyses support causal relationships between brain imaging-derived phenotypes and risk of psychiatric disorders 的文章。
研究背景
精神类疾病是一组脑功能紊乱的复杂疾病,会导致情感、认知和行为受到干扰和破坏。全球约有数亿人患有不同的精神障碍,被列为严重的公共卫生问题。近年来,脑影像学数据在脑疾病和功能的研究中受到广泛关注。以核磁共振成像为代表的脑影像技术,可用于活体无创定量评估人脑结构、连接和功能的特性。
虽然已有大量的观察性研究证据表明,精神疾病患者与健康正常人的脑影像表型存在显著差异,但脑影像学数据与精神障碍发病机制的因果关系尚不明确,探讨脑影像表型对精神疾病的因果作用具有重要的生物学和临床研究意义。
研究方法和结果
该研究基于大规模基因组数据,对常见的10种精神类疾病(包括注意力缺陷多动症、神经性厌食症、焦虑症、孤独症、双相情感障碍、抑郁症、强迫症、创伤后应激障碍、精神分裂症、抽动症)和587个关键的脑磁共振成像(MRI)结构表型进行了因果关系评估。
正向孟德尔随机化结果发现,脑白质纤维束的上额枕束的FA值和上放射冠的ICVF值、胼胝体内矢状层的MD值、第三脑室的体积等9个脑影像表型是精神分裂症、神经性厌食症和双相情感障碍的风险因素。进一步通过反向孟德尔随机化分析显示,发现精神分裂症的发生会导致额下回眶部的表面积和体积的增加。
该研究将基因组信息作为纽带,使脑影像表型和精神疾病联系起来,避免了观察性研究中由于药物或环境、生活方式等改变引起的样本检测数据偏差的缺点,确保了研究结果的稳健性。 参考技术A R语言中的随机效应模型更改代码如下:
fit <- summary(lm(b_out ~ -1 +b_exp, weights = 1/se_out^2))
代码里的b_out表示结局的beta值,b_exp表示暴露的beta值,se_out就是结局的标准误,se_out^2就代表结局beta值的方差,而模型中的-1表示的就是去除截距项。R语言里lm()函数表示拟合线性模型(linear model),summary()函数是用来汇总回归模型拟合的结果。关于R语言的相关信息,请参考往期推送R语言入门系列和R语言进阶系列。那么这次回归得出来的beta,se和P值就是MR分析的结果。
以上是关于mysql怎么把语言改成中文的主要内容,如果未能解决你的问题,请参考以下文章