差异表达基因分析:差异倍数(fold change), 差异的显著性(P-value)
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了差异表达基因分析:差异倍数(fold change), 差异的显著性(P-value)相关的知识,希望对你有一定的参考价值。
参考技术A Differential gene expression analysis:差异表达基因分析Differentially expressed gene (DEG):差异表达基因
差异表达分析是目前比较常用的识别疾病相关miRNA以及基因的方法,目前也有很多差异表达分析的方法,但比较简单也比较常用的是Fold change方法。
它的优点是计算简单直观,缺点是没有考虑到差异表达的统计显著性;通常以2倍差异为阈值,判断基因是否差异表达。Fold change的计算公式如下:
即用疾病样本的表达均值除以正常样本的表达均值。
差异表达分析的目的: 识别两个条件下表达差异显著的基因,即一个基因在两个条件中的表达水平,在排除各种偏差后,其差异具有统计学意义。我们利用一种比较常见的T检验(T-test)方法来寻找差异表达的miRNA。T检验的主要原理为:对每一个miRNA计算一个T统计量来衡量疾病与正常情况下miRNA表达的差异,然后根据t分布计算显著性p值来衡量这种差异的显著性,T统计量计算公式如下:
差异倍数(fold change)
fold change翻译过来就是倍数变化,假设A基因表达值为1,B表达值为3,那么B的表达就是A的3倍。一般我们都用count、TPM或FPKM来衡量基因表达水平,所以基因表达值肯定是非负数,那么fold change的取值就是(0, +∞).
为什么我们经常看到差异基因里负数代表下调、正数代表上调?因为我们用了log2 fold change。
当expr(A) < expr(B)时,B对A的fold change就大于1,log2 fold change就大于0(见下图),B相对A就是上调;
当expr(A) > expr(B)时,B对A的fold change就小于1,log2 fold change就小于0。
通常为了防止取log2时产生NA,我们会给表达值加1(或者一个极小的数),也就是log2(B+1) - log2(A+1). 【需要一点对数函数的基础知识】
为什么不直接用表达之差,差值接有正负啊?
假设A表达为1,B表达为8,C表达为64;直接用差值,B相对A就上调了7,C就相对B上调了56;用log2 fold change,B相对A就上调了3,C相对B也只上调了3.
通过测序观察我们发现,不同基因在细胞里的表达差异非常巨大,所以直接用差显然不合适, 用log2 fold change更能表示相对的变化趋势。
虽然大家都在用log2 fold change,但显然也是有缺点的:
一、到底是5到10的变化大,还是100到120的变化大?
二、5到10可能是由于技术误差导致的。所以当基因总的表达值很低时,log2 fold change的可信度就低了,尤其是在接近0的时候。
A disadvantage and serious risk of using fold change in this setting is that it is biased[7] and may misclassify differentially expressed genes with large differences (B − A) but small ratios (B/A), leading to poor identification of changes at high expression levels. Furthermore, when the denominator is close to zero, the ratio is not stable, and the fold change value can be disproportionately affected by measurement noise.
差异的显著性(P-value)
这就是统计学的范畴了,显著性就是根据假设检验算出来的。
假设检验首先必须要有假设,我们假设A和B的表达没有差异(H0,零假设),然后基于此假设,通过t test(以RT-PCR为例)算出我们观测到的A和B出现的概率,就得到了P-value, 如果P-value<0.05,那么说明小概率事件出现了,我们应该拒绝零假设,即A和B的表达不一样,即有显著差异。
显著性只能说明我们的数据之间具有统计学上的显著性,要看上调下调必须回去看差异倍数。
对于得到的显著性p值,我们需要进行多重检验校正(FDR),比较常用的是BH方法(Benjamini and Hochberg, 1995)。
这里只说了最基本的原理,真正的DESeq2等工具里面的算法肯定要复杂得多。
这张图对q-value(校正了的p-value)取了负log,相当于越显著,负log就越大,所以在火山图里,越外层的岩浆就越显著,差异也就越大。
只需要看懂DEG结果的可以就此止步,想深入了解的可以继续。
下面可以继续讨论的问题有:
1、RNA-seq基本分析流程/2、
2、DEG分析的常用算法/3、
3、常见DEG工具的方法介绍和相互比较
前言
做生物生理生化生信数据分析时,最常听到的肯定是“差异(表达)基因分析”了,从最开始的RT-PCR,到基因芯片microarray,再到RNA-seq,最后到现在的single cell RNA-seq,统统都在围绕着差异表达基因做文章。
(开个脑洞:再下一步应该会测细胞内特定空间内特定基因的动态表达水平了)
表达量 :我们假设基因转录表达形成的mRNA的数量反映了基因的活性,也会影响下游蛋白和代谢物的变化。我们关注的是 基因的表达 ,不是结构,也是不是isoform。
为什么差异基因分析这么流行?
一是中心法则得到了确立,基因表达是核心的一个环节,决定了下游的蛋白组和代谢组;
二是建库测序的普及,获取基因的表达水平变得容易。
在生物体内,基因的表达时刻都在动态变化,不一定服从均匀分布,在不同时间、发育程度、组织和环境刺激下,基因的表达肯定会发生变化。
差异基因分析主要应用在:
发育过程中关键基因的表达变化 - 发育研究
突变材料里什么核心基因的表达发生了变化 - 调控研究
细胞在受到药物处理后哪些基因的表达发生了变化 - 药物研发
目前我们对基因和转录组的了解到什么程度了?
基本的建库方法?建库直接决定了我们能测到什么序列,也决定了我们能做什么分析!
基因表达的normalization方法有哪些?
第一类错误、第二类错误是什么?
多重检验的校正?FDR?
10x流程解释
The mean UMI counts per cell of this gene in cluster i
The log2 fold-change of this gene's expression in cluster i relative to other clusters
The p-value denoting significance of this gene's expression in cluster i relative to other clusters, adjusted to account for the number of hypotheses (i.e. genes) being tested.
The differential expression analysis seeks to find, for each cluster, genes that are more highly expressed in that cluster relative to the rest of the sample. Here a differential expression test was performed between each cluster and the rest of the sample for each gene.
The Log2 fold-change (L2FC) is an estimate of the log2 ratio of expression in a cluster to that in all other cells. A value of 1.0 indicates 2-fold greater expression in the cluster of interest.
The p-value is a measure of the statistical significance of the expression difference and is based on a negative binomial test. The p-value reported here has been adjusted for multiple testing via the Benjamini-Hochberg procedure.
In this table you can click on a column to sort by that value. Also, in this table genes were filtered by (Mean UMI counts > 1.0) and the top N genes by L2FC for each cluster were retained. Genes with L2FC < 0 or adjusted p-value >= 0.10 were grayed out. The number of top genes shown per cluster, N, is set to limit the number of table entries shown to 10000; N=10000/K^2 where K is the number of clusters. N can range from 1 to 50. For the full table, please refer to the "differential_expression.csv" files produced by the pipeline.
不同单细胞DEG鉴定工具的比较
Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data
For data with a high level of multimodality, methods that consider the behavior of each individual gene, such as DESeq2, EMDomics, Monocle2, DEsingle, and SigEMD, show better TPRs. 这些工具敏感性高,就是说不会漏掉很多真的DEG,但是会包含很多假的DEG。
If the level of multimodality is low, however, SCDE, MAST, and edgeR can provide higher precision. 这些工具精准性很高,意味着得到的DEG里假的很少,所以会漏掉很多真的DEG,不会引入假的DEG。
time-course DEG analysis
Comparative analysis of differential gene expression tools for RNA sequencing time course data
参考:
Question: How to calculate "fold changes" in gene expression?
Exact Negative Binomial Test with edgeR
Differential gene expression analysis
R语言mRNA差异表达分析

上一篇文章介绍的是mRNA数据的整理,整理完就开始介绍的是如何用R语言来做差异表达分析,继续码字。
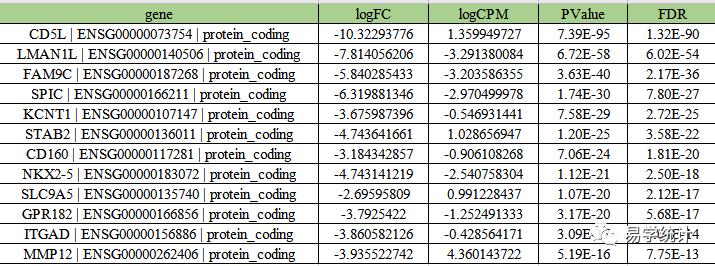
以上这个表格,在表达差异分析中很常见,第一列是基因名称。
第二列是基因的logFC值,FC是即fold change,表示实验组比上对照组的差异表达倍数。国内的一般默认取其对数绝对值大于2作为差异基因的筛选标准,这个阈值是可以根据实际情况进行调控。
第三列是logCPM,CPM是 Counts Per Million,是每个基因一个指标。
第四列是P值,其零假设是这个基因的表达在正常组织和癌症组织上是一样的,因此P<0.05可认为某个基因在两组间表达有显著差异。
第五列FDR是对P值的校正结果,一般使用FDR小于0.05。
火山图数据使用:FDR取对数和FC取对数,用来绘制火山图,并标注上调和下调基因。其中上调是指FC取对数大于某个正数,比如1,下调指的是FC取对数小于某个负数,比如-1.
热图数据使用:根据每个样本中每个基因的表达量绘制热图。其中每个小方格表示每个基因,颜色表示该基因表达量的大小。表达量越大,颜色越深(一般红色为上调,绿色为下调)
根据样本名称区分正常组织和癌症组织。首先加载上次处理后的数据集,上次用两种方法生成两个数据集,本次我们以第一个数据集为例子。
根据第一篇文章介绍01-09表示肿瘤样本,10-19表示正常对照组织。将癌症样本归为'tumor',正常对照组织归为'normal'。
setwd('~\下载数据\')load(file = "mRNA_exprSet.Rda")###metadata <- data.frame(names(mRNA_exprSet)[-1])for (i in 1:length(metadata[,1])) {num <- as.numeric(substring(metadata[i,1],14,15))if (num %in% seq(1,9)) {metadata[i,2] <- "tumor"}if (num %in% seq(10,29)) {metadata[i,2] <- "normal"}}
打开基因数据这个文件夹 可以看到里面有182个文件夹,每一个文件夹里面还有1个压缩文件。这里面其实就是下载的182个样本,每个样本的信息单独保存在一个文件夹。这种数据不能直接拿来做分析,因此先要合并。
mycounts <- mRNA_exprSetmycounts=as.matrix(mycounts) ##DGEList对象要求矩阵格式rownames(mycounts)=mycounts[,1]exp=mycounts[,2:ncol(mycounts)]dimnames=list(rownames(exp),colnames(exp))data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)data=avereps(data) ##如果有重名的基因,就自动取平均值,方便后面的分析data=data[rowMeans(data)>1,] ##这个基因在所有的样品上基本都不表达或者是低表达,就删除。
预处理前是19668个基因,删除在所有样品上基本不表达或者低表达的基因,剩下17922个基因。
也可以采用cpm校准化的基因表达量作为过滤低表达基因的标准。
group <- as.vector(metadata$sample) #按照癌症和正常样品数目修改design <- model.matrix(~group) ##变成一个实验设计对象y <- DGEList(counts=data,group=group) ##变成edgr包可以识别的数据结构,data需要是矩阵格式
edgeR这个包将数据存储在DGEList对象中,需要指定的参数包括counts 和 group。其中表达矩阵存储在$counts中,样本分组信息等存储在$samples中。
如果采用cpm过滤基因,R代码操作如下:
# cpm normalizationcountsPerMillion <- cpm(y)countCheck <- countsPerMillion > 1keep <- which(rowSums(countCheck) >= 2)<- y[keep,]##16889 182
①构建完DGEList对象,做因子校正
y <- calcNormFactors(y) ##对因子进行校正edgeR默认使用TMM(trimmed mean of M-values)进行normalization,以最大程度地缩小样本间基因表达量的log-fold change。这是因为TMM 法认为样本间大部分的基因都没有发生差异表达,而那些真正差异表达的基因并不会受到normalization的严重影响。如此一来,便将那些由于测序引起的差异表达基因的表达量给校正了,消除了一部分的假阳性。
为什么要做Normalization呢?
在差异分析中,我们常常更关注的是相对表达量的变化,例如处理组的A基因表达量相对于对照组的而言是上调还是下调了。而基因表达量的定量准确性则在差异分析中不太重要,因此,在进行差异分析时,像RPKM/FPKM这种对转录本长度进行normalization方法是并不常用,也是没有必要的。
在常规的RNA-seq中,影响基因表达量更大的技术因素往往是测序深度以及有效文库大小(effective libraries size)。这也是一般的差异分析软件会进行normalize的部分
y <- estimateCommonDisp(y) ##为所有基因都计算同样的dispersiony <- estimateTrendedDisp(y) ##根据不同基因的均值--方差之间的关系来计算dispersion,并拟合一个trended modely <- estimateTagwiseDisp(y) ##计算每个基因的dispersion,并利用经验性贝叶斯方法(empirical bayes)压缩至Trend Didspersion。##个人认为这一项相当于GLM中每个基因的beta值plotBCV(y)
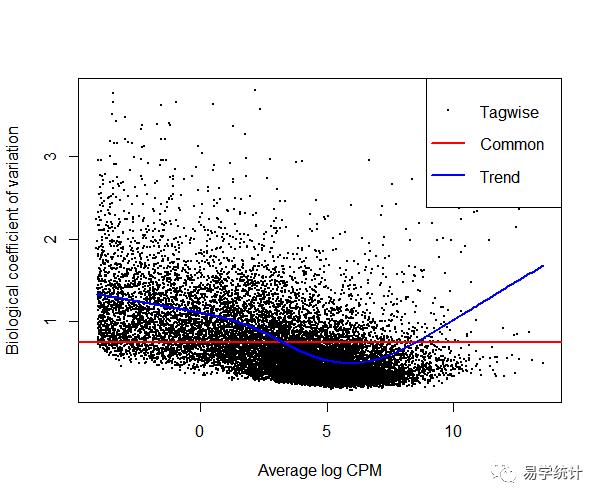
③离散值估计的结果解释:
关于dispersion estimation
一般而言,样本间的变异系数(coefficient of variance,CV)是由两部分组成的,一是技术差异(Technical CV),另一个是生物学差异(Biological coefficient of variance,BCV)。前者是会随着测序通量的提升而消失的,而后者则是样本间真实存在的差异。所以,若一个基因的BCV值在样本间比较大的话,可认为这是个差异表达基因。而edgeR正是通过估计dispersion来估计BCV,进而拟合出线性回归模型的参数。
BCV是负二项分布的平方根。 该图显示BCV估计值和趋势估计值。
这里引出了负二项分布,为什么用这个分布?
在基因组学中,基因表达的reads数,可以看作一个计数的过程,我们可以把采样自某个基因的reads数量建模为泊松分布,并利用泊松回归估计具体的数量。
然而,对于同一个基因来说,通常var比mean要大(比如进行了3次生物学重复实验,假设测了3只同样疾病状态的小鼠;注意,生物重复不是技术重复;这3次得到某基因的reads counts,一般来讲方差大于其均值),特别是对于高表达的基因。因此,负二项回归要更合适一点。
Poisson分布与负二项分布有什么关系呢?Poisson分布中,设定μ是常数,当μ不是常数,而是一个随机变量,且服从γ分布时,此时复合Poisson分布就是负二项分布。
而负二项分布(NB)需要均值和离散值两个参数。edgeR对每个基因都估测一个经验贝叶斯稳健离散值(mpirical Bayes moderated dispersion),还有一个公共离散值(common dispersion,所有基因的经验贝叶斯稳健离散值的均值)以及一个趋势离散值,见上代码注释。

④:显著性差异检验:
et <- exactTest(y,pair = c("normal","tumor")) #检验两组时间的变异系数## ##类似配对T检验ordered_tags <- topTags(et, n=100000)##P值的校正 采用BH方法
这个执行完就是出来文章开头讲的表格,差异表达的工作已经完成,后面的工作就是基于这个表格做一些基因的筛选和图片的绘制。
allDiff=ordered_tags$tableallDiff=allDiff[is.na(allDiff$FDR)==FALSE,]diff=allDiff ##提取所有基因差异分析后的结果,见下表1newData=y$pseudo.counts ##提取校准后表达的数据,见下表2
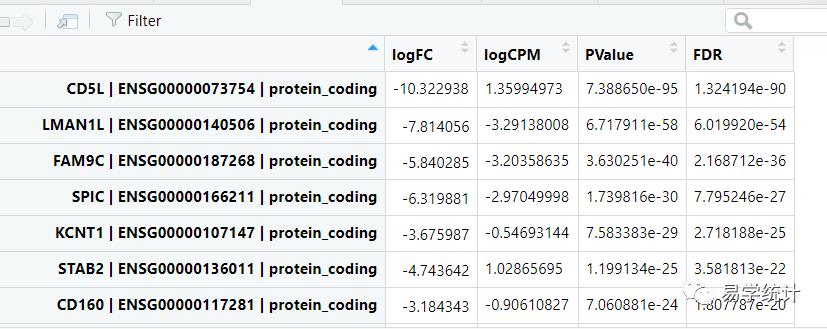
②筛选
###定义两个筛选条件foldChange=1 #差异在2倍以上padj=0.05 ##校正后的P值小于0.05###筛选P值小于0.05且logFC的绝对值大于1的基因diffSig = diff[(diff$FDR < padj & (diff$logFC>foldChange | diff$logFC<(-foldChange))),]write.table(diffSig, file="diffSig.xls",sep=" ",quote=F)## 662 4###显示上调基因###筛选P值小于0.05且logFC的大于1的基因diffUp = diff[(diff$FDR < padj & (diff$logFC>foldChange)),]write.table(diffUp, file="up.xls",sep=" ",quote=F)## 36 4##显示下调基因###筛选P值小于0.05且logFC的小于-1的基因diffDown = diff[(diff$FDR < padj & (diff$logFC<(-foldChange))),]write.table(diffDown, file="down.xls",sep=" ",quote=F)## 626 4###输出所有基因校正后的表达normalizeExp=rbind(id=colnames(newData),newData)write.table(normalizeExp,file="normalizeExp.txt",sep=" ",quote=F,col.names=F)#输出差异基因校正后的表达diffExp=rbind(id=colnames(newData),newData[rownames(diffSig),])write.table(diffExp,file="diffmRNAExp.txt",sep=" ",quote=F,col.names=F)heatmapData <- newData[rownames(diffSig),]
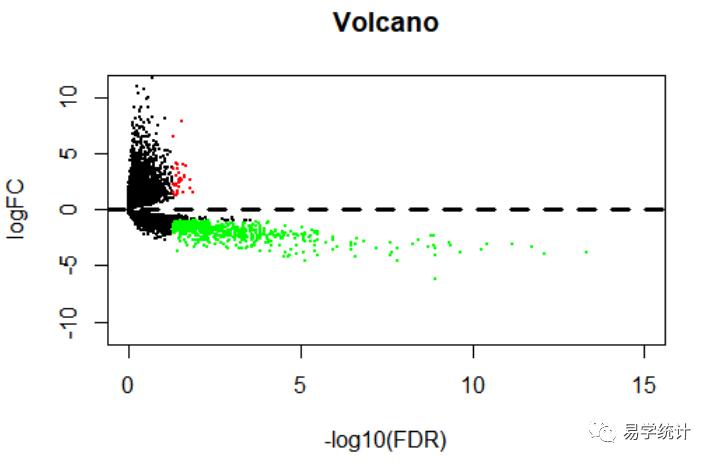
火山图用途?
下图1中,火山图可反映总体基因的表达情况,横坐标代表log2(Fold Change),纵坐标表示-log10(P值),值越大表示差异越显著,每个点代表一个基因,绿色点是下调2倍以上的基因,红色点是上调2倍以上的基因。处于二者之间的标记为黑色。
下面我们看一下火山图的绘制。
pdf(file="vol.pdf")xMax=max(-log10(allDiff$FDR))+1yMax=12plot(-log10(allDiff$FDR), allDiff$logFC, xlab="-log10(FDR)",ylab="logFC",main="Volcano", xlim=c(0,xMax),ylim=c(-yMax,yMax),yaxs="i",pch=20, cex=0.4)diffSub=allDiff[allDiff$FDR<padj & allDiff$logFC>foldChange,]points(-log10(diffSub$FDR), diffSub$logFC, pch=20, col="red",cex=0.4)diffSub=allDiff[allDiff$FDR<padj & allDiff$logFC<(-foldChange),]points(-log10(diffSub$FDR), diffSub$logFC, pch=20, col="green",cex=0.4)abline(h=0,lty=2,lwd=3)dev.off()
聚类热图用途?
横坐标代表样本聚类,一列代表一个样本,聚类基于样本间基因表达的相似性,样本间基因表达越接近,靠的越近,以此类推。
纵坐标代表基因聚类,一行代表一个基因,聚类基于基因在样本中表达的相似性,基因在样本中表达越接近,靠的越近,以此类推。
色阶代表基因表达丰度,越红代表上调得越明显,越绿代表下调得越明显。这图一般做展示数据用,看上去直观点。
hmExp=log10(heatmapData+0.001)library('gplots')hmMat=as.matrix(hmExp)pdf(file="heatmap.pdf",width=60,height=90)par(oma=c(10,3,3,7))heatmap.2(hmMat,col='greenred',trace="none")dev.off()
本次完成基因差异的分析和两个常见图的绘制,下期分享共表达。
作者介绍:医疗大数据统计分析师,擅长R语言。
欢迎各位在后台留言,恳请斧正!
更多阅读
长按二维码
易学统计
以上是关于差异表达基因分析:差异倍数(fold change), 差异的显著性(P-value)的主要内容,如果未能解决你的问题,请参考以下文章