GEO数据挖掘-第二期-三阴性乳腺癌(TNBC)
Posted 生信草堂
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了GEO数据挖掘-第二期-三阴性乳腺癌(TNBC)相关的知识,希望对你有一定的参考价值。
这个数据集一共有三篇文章对他进行了数据挖掘:
PMID: 25208879
PMID: 26921331
PMID: 30175120
我们今天实践最后一个。
文章标题
Identification of Key Genes and Pathways in Triple-Negative Breast Cancer by Integrated Bioinformatics Analysis
关键词
三阴性乳腺癌
疾病:三阴性乳腺癌
不表达下面的受体
1.estrogen receptor (ER)
2.progesterone receptor(PR)
3.human epidermal growth factor receptor 2 (Her2)
◆ ◆ ◆ ◆ ◆
GEO数据库编号:GSE76275
实验设计
实验组:198个三阴性乳腺癌肿瘤样本
对照组:67非三阴性乳腺癌肿瘤样本
◆ ◆ ◆ ◆ ◆
GEO数据挖掘过程
比较198个三阴性乳腺癌肿瘤样本和67非三阴性乳腺癌肿瘤样本
library( "GEOquery" )
## 取表达矩阵和样本信息表
{
gset = gset[[1]]
exprSet = exprs( gset )
pdata = pData( gset )
chl = length( colnames( pdata ) )
group_list = as.character( pdata[, 67] )
}
dim( exprSet )
exprSet[ 1:5, 1:5 ]
table( group_list )
## 取出研究样本
{
not_TN_expr = exprSet[ , grep( "not TN", group_list )]
TN_expr = exprSet[ , !(colnames(exprSet) %in% colnames(not_TN_expr)) ]
exprSet=cbind(not_TN_expr, TN_expr)
}
## 样本分组
{
group_list = c(rep( 'not_TN', ncol( not_TN_expr ) ),
rep( 'TN', ncol( TN_expr ) ) )
}
dim( exprSet )
exprSet[ 1:5, 1:5 ]
table( group_list )
save( exprSet, group_list, file = 'exprSet_by_group.Rdata')
GPL570的一个探针对应了多个基因。
## 筛选探针
GPL = gset@featureData@data
colnames( GPL )
View( GPL )
ids = GPL[ ,c( 1, 11 ) ]
ids = ids[ ids[ , 2 ] != '' , ]
## 一个探针对应多个基因
library("plyr")
a<-strsplit(as.character(ids[,2]), " /// ")
tmp <- mapply( cbind, ids[,1], a )
df <- ldply (tmp, data.frame)
ID2gene = df[,2:3]
colnames( ID2gene ) = c( "id", "gene" )
dim(ID2gene)
save(ID2gene, file = 'ID2gene.Rdata')
这个数据集不需要进行log处理。
取相同基因的表达数据的最大值这一步,运行时间长,大家可以想下有没有优雅的代码解决这个问题。
{
exprSet = exprSet[ rownames(exprSet) %in% ID2gene[ , 1 ], ]
ID2gene = ID2gene[ match(rownames(exprSet), ID2gene[ , 1 ] ), ]
}
dim( exprSet )
dim( ID2gene )
tail( sort( table( ID2gene[ , 2 ] ) ), n = 12L )
## 相同基因的表达数据取最大值
{
MAX = by( exprSet, ID2gene[ , 2 ],
function(x) rownames(x)[ which.max( rowMeans(x) ) ] )
MAX = as.character(MAX)
exprSet = exprSet[ rownames(exprSet) %in% MAX , ]
rownames( exprSet ) = ID2gene[ match( rownames( exprSet ), ID2gene[ , 1 ] ), 2 ]
}
dim(exprSet)
exprSet[1:5,1:5]
save(exprSet, group_list, file = 'final_exprSet.Rdata')
这步结束我们就得到了最后的数据集。
data = as.data.frame( t( exprSet ) )
data$group = group_list
png( 'pca_plot.png', res=100 )
autoplot( prcomp( data[ , 1:( ncol( data ) - 1 ) ] ), data = data, colour = 'group') + theme_bw()
dev.off()
library( "limma" )
{
design <- model.matrix( ~0 + factor( group_list ) )
colnames( design ) = levels( factor( group_list ) )
rownames( design ) = colnames( exprSet )
}
design
contrast.matrix <- makeContrasts( "TN-not_TN", levels = design )
contrast.matrix
load( "./ID2gene.Rdata" )
{
fit <- lmFit( exprSet, design )
fit2 <- contrasts.fit( fit, contrast.matrix )
fit2 <- eBayes( fit2 )
nrDEG = topTable( fit2, coef = 1, n = Inf )
write.table( nrDEG, file = "nrDEG.out")
}
head(nrDEG)
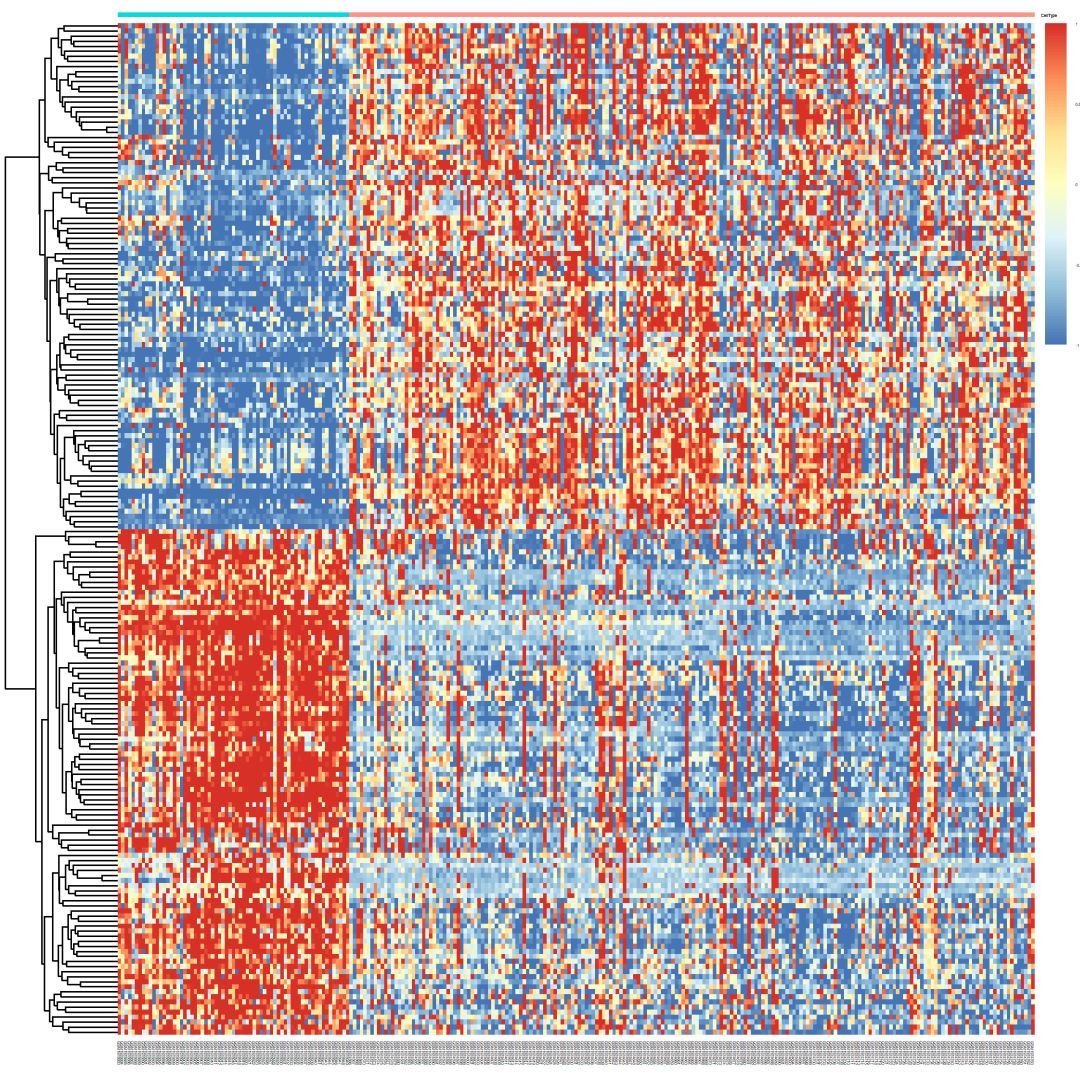
## heatmap
library( "pheatmap" )
{
nrDEG_Z = nrDEG[ order( nrDEG$logFC ), ]
nrDEG_F = nrDEG[ order( -nrDEG$logFC ), ]
choose_gene = c( rownames( nrDEG_Z )[1:100], rownames( nrDEG_F )[1:100] )
choose_matrix = exprSet[ choose_gene, ]
choose_matrix = t( scale( t( choose_matrix ) ) )
choose_matrix[choose_matrix > 1] = 1
choose_matrix[choose_matrix < -1] = -1
annotation_col = data.frame( CellType = factor( group_list ) )
rownames( annotation_col ) = colnames( exprSet )
pheatmap( fontsize = 2, choose_matrix, annotation_col = annotation_col, show_rownames = F,
annotation_legend = F, cluster_cols = F, filename = "heatmap.png")
}
## volcano plot
library( "ggplot2" )
logFC_cutoff <- with( nrDEG, mean( abs( logFC ) ) + 2 * sd( abs( logFC ) ) )
logFC_cutoff
logFC_cutoff = 1.5
{
nrDEG$change = as.factor( ifelse( nrDEG$P.Value < 0.01 & abs(nrDEG$logFC) > logFC_cutoff,
ifelse( nrDEG$logFC > logFC_cutoff , 'UP', 'DOWN' ), 'NOT' ) )
save( nrDEG, file = "nrDEG.Rdata" )
this_tile <- paste0( 'Cutoff for logFC is ', round( logFC_cutoff, 3 ),
' The number of up gene is ', nrow(nrDEG[ nrDEG$change =='UP', ] ),
' The number of down gene is ', nrow(nrDEG[ nrDEG$change =='DOWN', ] ) )
volcano = ggplot(data = nrDEG, aes( x = logFC, y = -log10(P.Value), color = change)) +
geom_point( alpha = 0.4, size = 1.75) +
theme_set( theme_set( theme_bw( base_size = 15 ) ) ) +
xlab( "log2 fold change" ) + ylab( "-log10 p-value" ) +
ggtitle( this_tile ) + theme( plot.title = element_text( size = 15, hjust = 0.5)) +
scale_colour_manual( values = c('blue','black','red') )
print( volcano )
ggsave( volcano, filename = 'volcano.png' )
dev.off()
}
library( "clusterProfiler" )
library( "org.Hs.eg.db" )
df <- bitr( rownames( nrDEG ), fromType = "SYMBOL", toType = c( "ENTREZID" ), OrgDb = org.Hs.eg.db )
head( df )
{
nrDEG$SYMBOL = rownames( nrDEG )
nrDEG = merge( nrDEG, df, by='SYMBOL' )
}
head( nrDEG )
{
gene_up = nrDEG[ nrDEG$change == 'UP', 'ENTREZID' ]
gene_down = nrDEG[ nrDEG$change == 'DOWN', 'ENTREZID' ]
gene_diff = c( gene_up, gene_down )
gene_all = as.character(nrDEG[ ,'ENTREZID'] )
}
{
geneList = nrDEG$logFC
names( geneList ) = nrDEG$ENTREZID
geneList = sort( geneList, decreasing = T )
}
{
## KEGG pathway analysis
kk.up <- enrichKEGG( gene = gene_up ,
organism = 'hsa' ,
universe = gene_all ,
pvalueCutoff = 0.8 ,
qvalueCutoff = 0.8 )
kk.down <- enrichKEGG( gene = gene_down ,
organism = 'hsa' ,
universe = gene_all ,
pvalueCutoff = 0.8 ,
qvalueCutoff = 0.8 )
}
head( kk.up )[ ,1:6 ]
head( kk.down )[ ,1:6 ]
library( "ggplot2" )
{
kegg_down_dt <- as.data.frame( kk.down )
kegg_up_dt <- as.data.frame( kk.up )
down_kegg <- kegg_down_dt[ kegg_down_dt$pvalue < 0.05, ]
down_kegg$group = -1
up_kegg <- kegg_up_dt[ kegg_up_dt$pvalue < 0.05, ]
up_kegg$group = 1
dat = rbind( up_kegg, down_kegg )
dat$pvalue = -log10( dat$pvalue )
dat$pvalue = dat$pvalue * dat$group
dat = dat[ order( dat$pvalue, decreasing = F ), ]
g_kegg <- ggplot( dat,
aes(x = reorder( Description, order( pvalue, decreasing=F ) ), y = pvalue, fill = group)) +
geom_bar( stat = "identity" ) +
scale_fill_gradient( low = "blue", high = "red", guide = FALSE ) +
scale_x_discrete( name = "Pathway names" ) +
scale_y_continuous( name = "log10P-value" ) +
coord_flip() + theme_bw() + theme( plot.title = element_text( hjust = 0.5 ) ) +
ggtitle( "Pathway Enrichment" )
print( g_kegg )
ggsave( g_kegg, filename = 'kegg_up_down.png' )
}
◆ ◆ ◆ ◆ ◆
以上是关于GEO数据挖掘-第二期-三阴性乳腺癌(TNBC)的主要内容,如果未能解决你的问题,请参考以下文章