R语言实现流式细胞数据分析
Posted R语言交流中心
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言实现流式细胞数据分析相关的知识,希望对你有一定的参考价值。
流式细胞术通过光学检测系统快速检测多参数的细胞流。许多因素使得流式细胞术能够成功和广泛的应用,比如检测速度(能够允许大量的细胞被检测),高度的准确性和分辨率,低成本。此外,流式细胞术还是一种非破坏性技术,可以分选出活细胞用于后续分析。能够分析和分选单个细胞的能力使流式细胞术在生物学和医学领域有非常广泛的应用。在免疫学中,流式细胞术用来鉴定和量化免疫细胞亚群,因此可以监控病人的免疫状态,通过比较不同的病人组也可以找出生物标志物。具体的原理:一定波长的激光束直接照射到高压驱动的液流,产生的光信号被多个接收器接受,一个是机关束直线方向上接受的前向角散射光信号。其他是在激光束垂直方向上接受的光信号,包括侧向角散射光信号和荧光信号,这些光信号被相应的接受器接受后,根据接收信号的强弱就能反应出细胞的物理和化学特征。
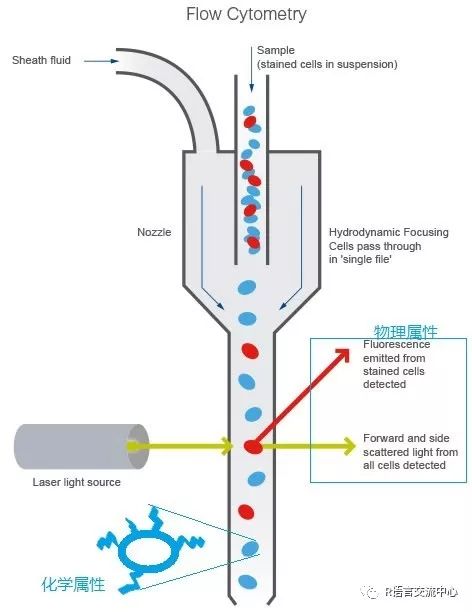
其中主要的信号包括:前向角散射信号(FSC)反映细胞的大小;侧向角散射信号(SSC)反映细胞颗粒度值越大,细胞内颗粒结构越复杂,质量越大;荧光信号(FL)反映胞内、胞外染色情况。接下来我们看下在R语言如何进行整个的分析流程。首先我们需要安装其几个重要的包:
##数据源:BiocManager::install("flowWorkspaceData")##数据读取和质控:BiocManager::install("flowCore")BiocManager::install("flowClean")##批量标准化:BiocManager::install("flowStats")##自动圈门包:BiocManager::install("openCyto")BiocManager::install("flowDensity")##结果的可视化:BiocManager::install("ggcyto")BiocManager::install("flowViz")
以上的包便可以完成流式的整个分析过程,接下来我们就看下如何实现整个分析流程:
1. 载入数据
library(flowWorkspaceData)dataDir <-system.file("extdata",package="flowWorkspaceData")library(flowCore)file.name <-system.file("extdata","0877408774.B08",package="flowCore")fcs.dir <-system.file("extdata","compdata","data",package="flowCore")
2. 读取数据及可视化
首先我们看下flowCore包中的读取数据的函数read.FCS和read.flowSet:


其中主要的参数:
Transformation 主要是指的原始数据的转化,默认是线性转化,一般我不需要转化直接设为FALSE
Which.lines 主要是指的读取的范围,也就是行数eg:which.lines=2:5
实例:
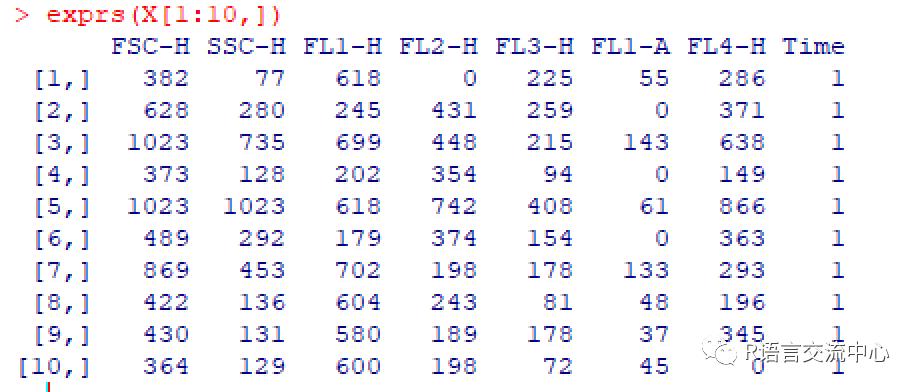
X=read.FCS(file.name, transformation=FALSE)Y=read.FCS(paste(dataDir,'/CytoTrol_CytoTrol_1.fcs',sep=''),transformation=FALSE)exprs(X[1:10,])##获取对应的信号的值parameters(X)$name###获取X中的变量keyword(X) ###获取文件的关键信息
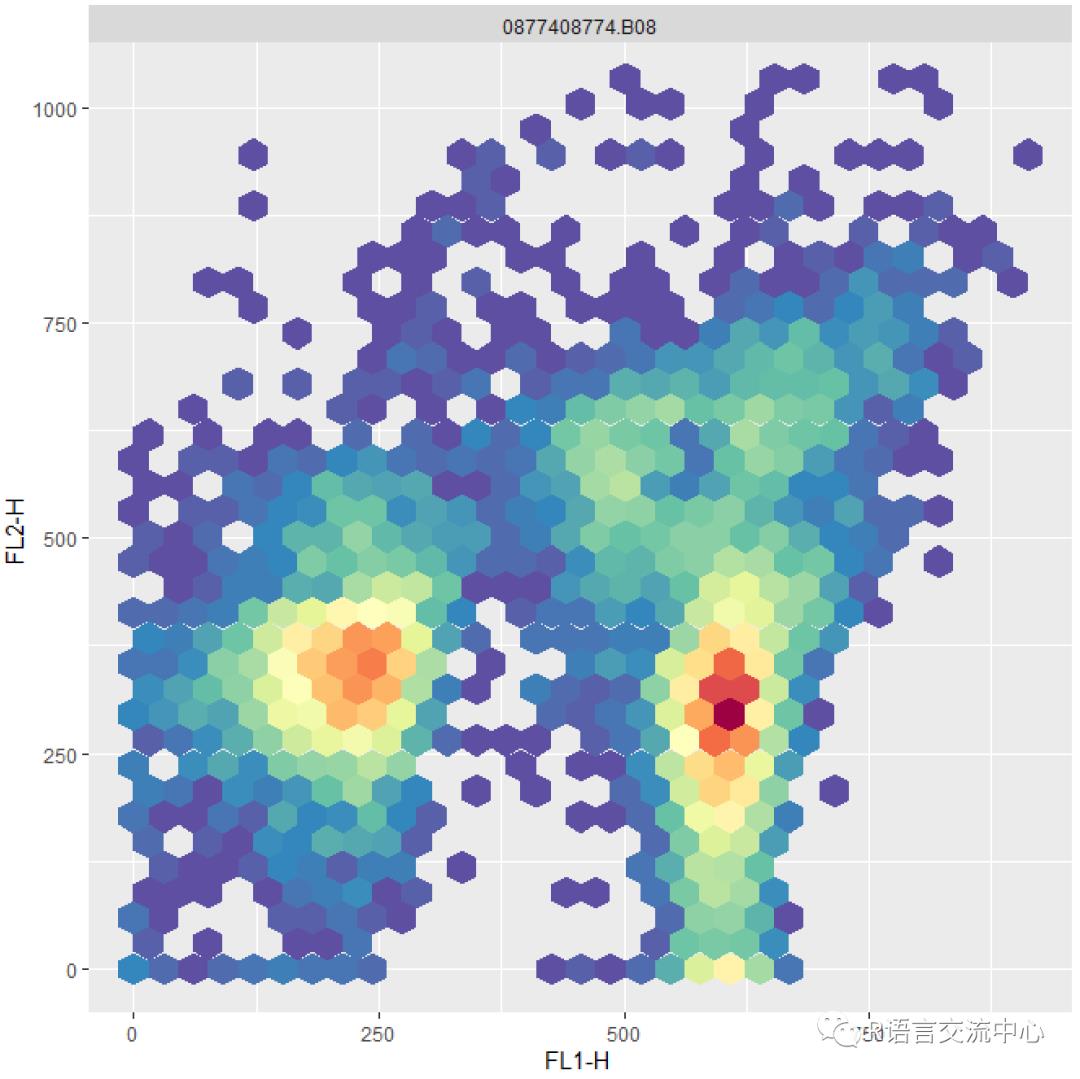
library(ggcyto)autoplot(X, "FL1-H","FL2-H")
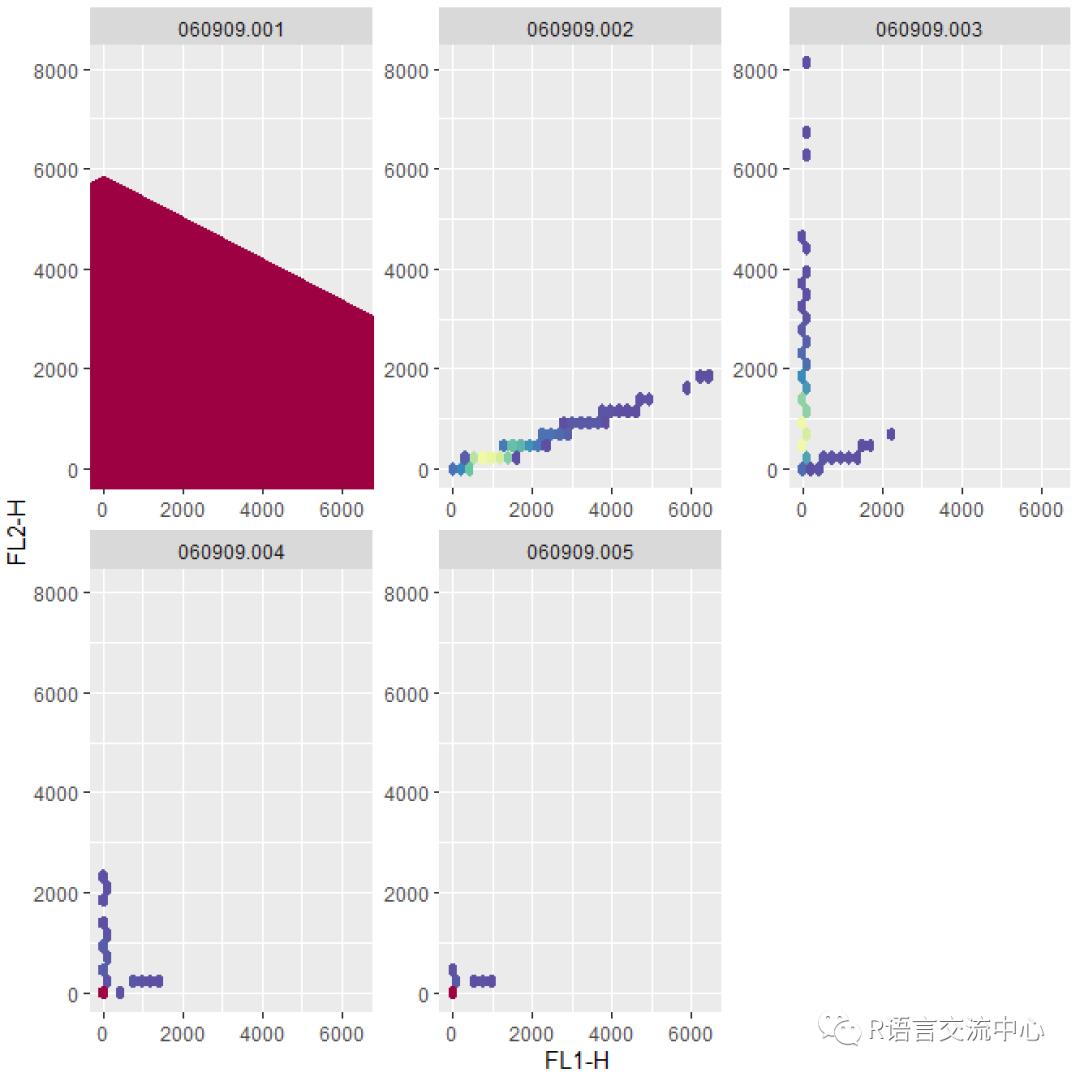
fs <- read.flowSet(path = fcs.dir)sampleNames(fs)##查看样本编号信息,多批实验时可用。exprs(fs[[1]][1:12,])##获取数据autoplot(fs, "FL1-H","FL2-H")
那如果我们的荧光素1对应的通道可以检测其荧光信号,那么其它的通道也会产生荧光素1的信号,那这些信号就成为溢出,组成的荧光信号值的矩阵也就是溢出矩阵,在flowCore中作者提到了对溢出矩阵的处理方法:
首先获取溢出矩阵:
fcsfiles <- list.files(pattern ="CytoTrol",system.file("extdata",package="flowWorkspaceData"),full = TRUE)fs <- read.flowSet(fcsfiles)x <- fs[[1]]comp_list <- spillover(x)comp_list

接下来我们看下如何进行标准化处理:
comp <- comp_list[[1]]x_comp <- compensate(x, comp)##单批数据处理comp <- fsApply(fs, function(x)spillover(x)[[1]], simplify=FALSE) ###多批处理fs_comp <- compensate(fs, comp)
library(gridExtra)transList <- estimateLogicle(x,c("V450-A","V545-A"))p1 <- autoplot(transform(x, transList),"V450-A", "V545-A") +ggtitle("Before")p2 <- autoplot(transform(x_comp,transList), "V450-A", "V545-A")+ggtitle("Before")grid.arrange(as.ggplot(p1), as.ggplot(p2),ncol = 2)
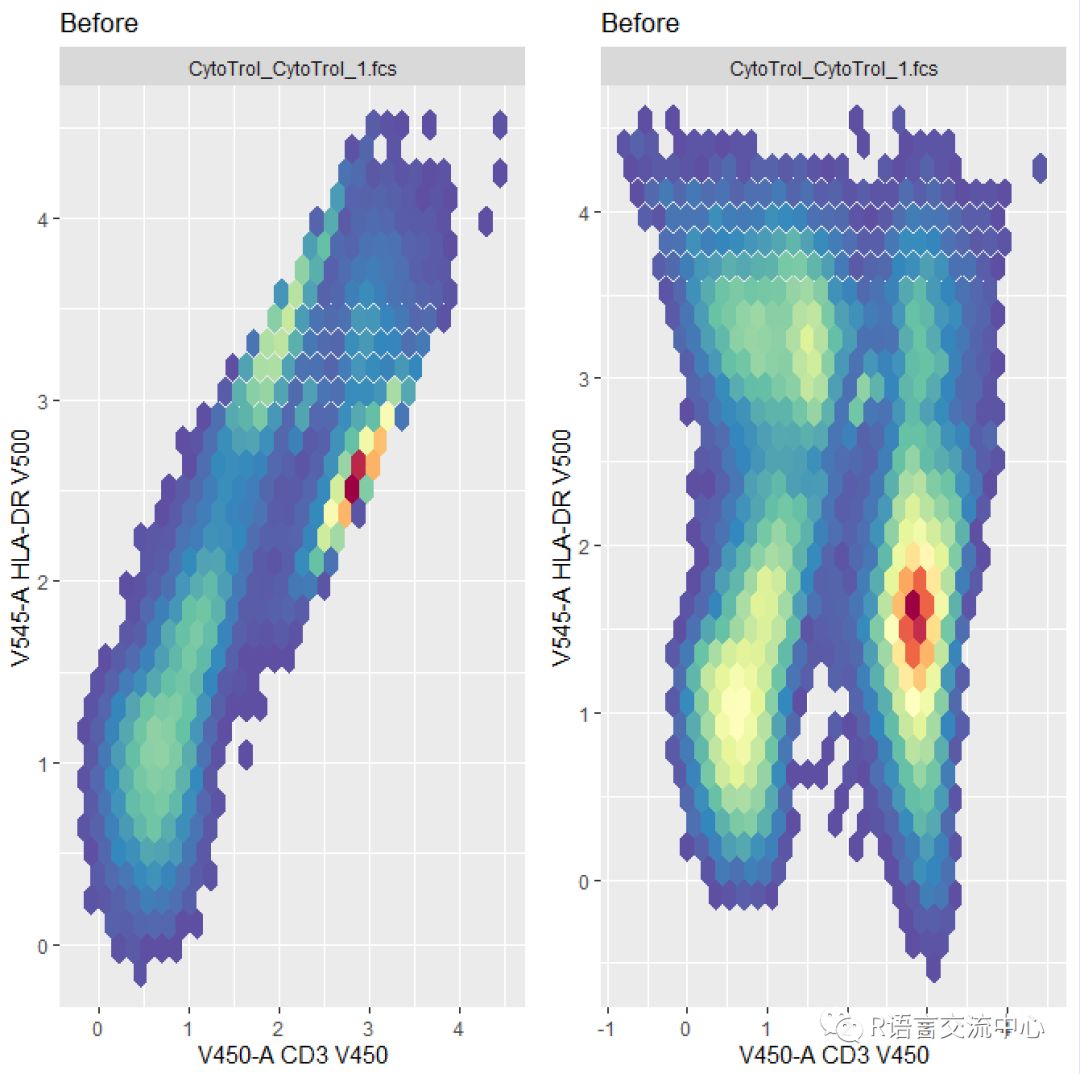
从上图我们可以看出在经过溢出矩阵处理后的效果,可以更好的将数据的差异分开。
3. 质量控制
我们主要是看下flowClean中clean函数进行对flowdata类型的数据进行质量控制并获得质控之后的数据,当然这个函数有一个缺点,那就是细胞数必须大于30000,不然没法进行质控:
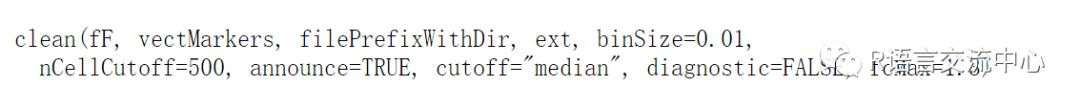
主要的参数:
vectMarkers 指的要进行标准化的列。e.g. 'FSC-A'
nCellCutoff 指一个群体最小的细胞数。
Cutoff 指的阈值,可以是0-1的阈值,当然也可以是大于1的荧光值。
filePrefixWithDir 输出的文件路径或者文件名
Ext 文件扩展名
实例:
library(flowClean)library(flowViz)data(synPerturbed)synPerturbed.c <- clean(synPerturbed,vectMarkers=c(5:16),filePrefixWithDir="sample_out",ext="fcs", diagnostic=TRUE)library(grid)library(gridExtra)lgcl <- estimateLogicle(synPerturbed.c,unname(parameters(synPerturbed.c)$name[5:16])) ##获取要进行转化的列对应的名称。synPerturbed.cl <-transform(synPerturbed.c, lgcl) ##转化后的数据集p1 <- xyplot(`<V705-A>` ~ `Time`,data=synPerturbed.cl, abs=TRUE, smooth=FALSE, alpha=0.5, xlim=c(0, 100))p2 <- xyplot(`GoodVsBad` ~ `Time`,data=synPerturbed.cl, abs=TRUE, smooth=FALSE, alpha=0.5, xlim=c(0, 100),ylim=c(0, 20000))rg <-rectangleGate(filterId="gvb", list("GoodVsBad"=c(0, 9999)))idx <- filter(synPerturbed.cl, rg) ###获取过滤后的结果synPerturbed.clean <-Subset(synPerturbed.cl, idx)p3 <- xyplot(`<V705-A>` ~ `Time`,data=synPerturbed.clean, abs=TRUE, smooth=FALSE, alpha=0.5, xlim=c(0, 100))grid.arrange(p1, p2, p3, ncol=3)
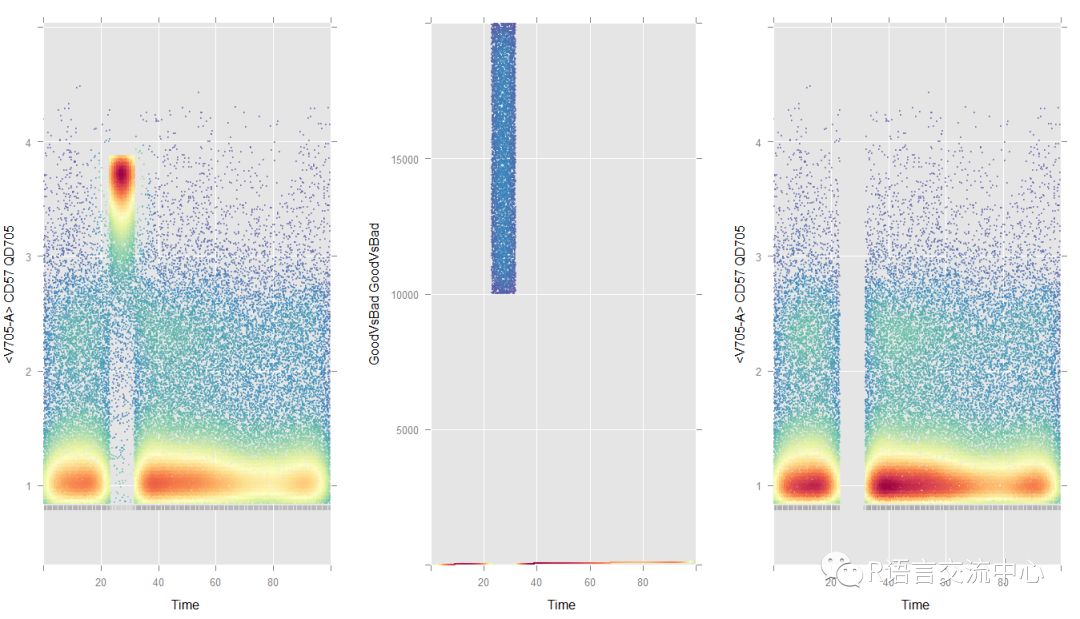
从上图我们可以看出在过滤后,数据随着时间的增加会清晰的分开。
我们看下flowCore包中的数据的标准化需要用到函数transfom,我们直接看一个对数转换的实例:
fs <-read.flowSet(path=system.file("extdata", "compdata","data", package="flowCore"), name.keyword="SAMPLEID", phenoData=list(name="SAMPLE ID",Filename="$FIL"))autoplot(transform(fs[[1]],`FL1-H`=log(`FL1-H`), `FL2-H`=log(`FL2-H`) ),"FL1-H","FL2-H")
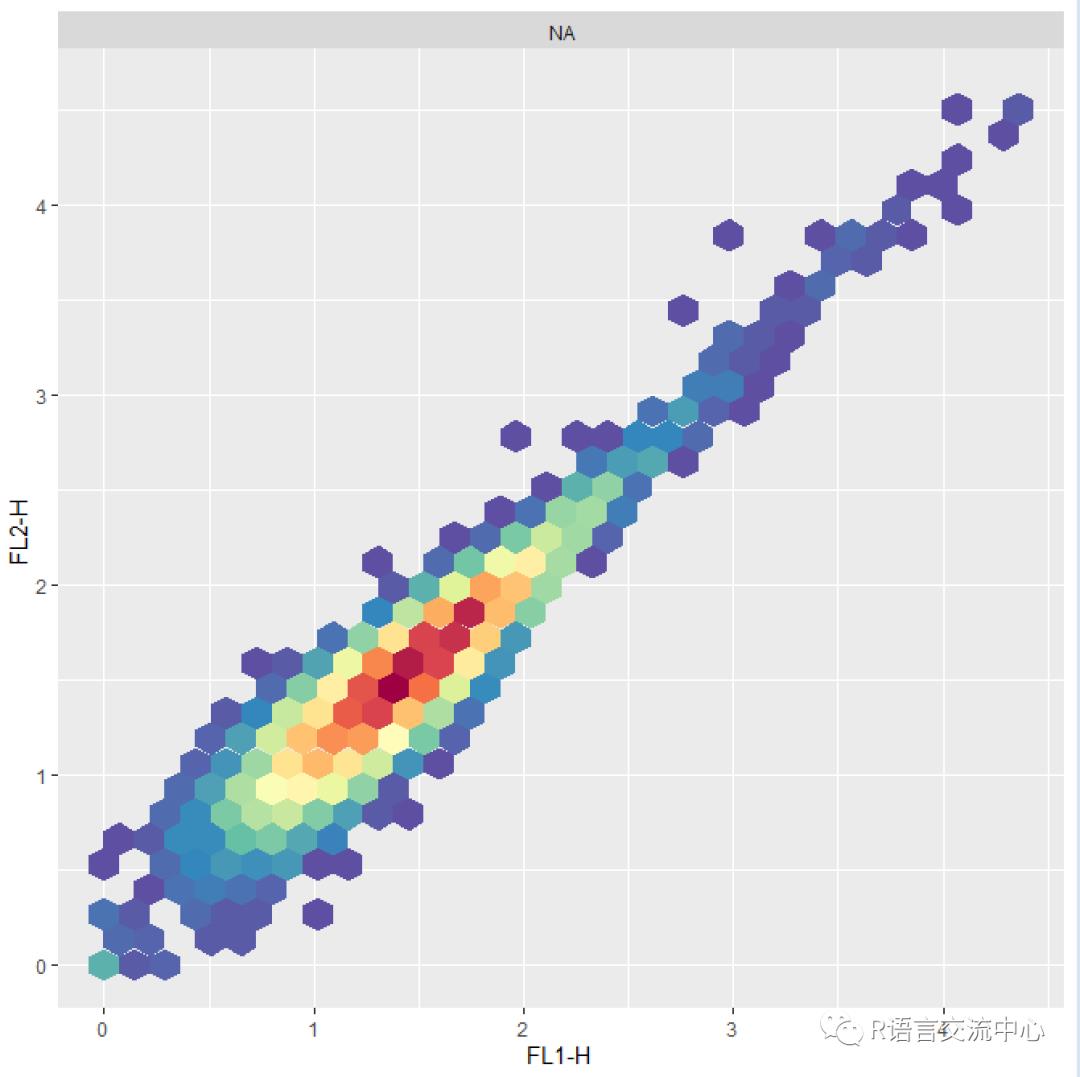
当然我们的标准化形式不止这一种,还有下面的几种:

那这些的使用就比较复杂了,我们直接上实例:
aTrans <-truncateTransform("truncate at 1", a=1) ##构造参数myTrans <- transformList('FL1-H',aTrans)transform(fs, myTrans)
对于一个数据集基本的流程如上,那么如果多个数据集我们需要用到flowStats,此包主要用于数据的标准化和自动圈门。那么我们看下具体的应用:
library(flowStats)fcs.dir <-system.file("extdata", "compdata","data",package="flowCore")frames <- lapply(dir(fcs.dir,full.names=TRUE), read.FCS)names(frames) <-c("UNSTAINED", "FL1-H", "FL2-H","FL4-H", "FL3-H")frames <- as(frames,"flowSet")##构建flowset,当然可以直接用flowCore读取生成flowsetcomp <- spillover(frames,unstained="UNSTAINED", patt = "-H",fsc = "FSC-H",ssc = "SSC-H",stain_match = "ordered")
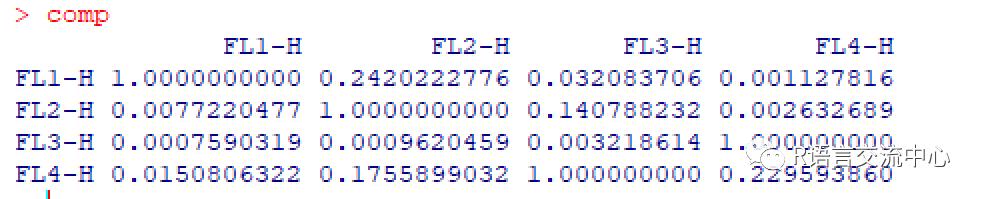
当然这里是不考虑通道的对应性时stain_match为ordered,当需要一一对应时则需要另一个参数regexpr.
comp <- spillover(frames,unstained="UNSTAINED", patt = "-H", fsc ="FSC-H", ssc = "SSC-H", stain_match = "regexpr")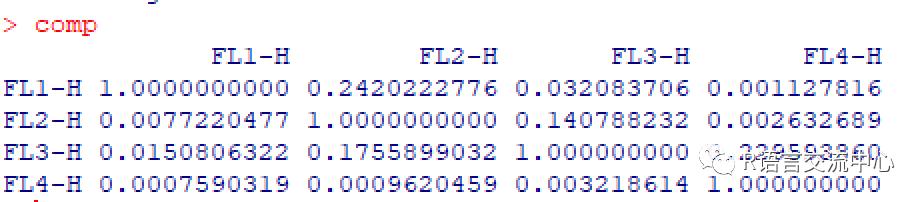
那么除了上面的方法,还提供了spillover_match函数,可以直接对目录下的文件进行匹配:
comp_match <-system.file("extdata", "compdata", "comp_match",package="flowCore")control_path <-system.file("extdata", "compdata", "data",package="flowCore")matched_fs <-spillover_match(path=control_path, fsc = "FSC-H", ssc ="SSC-H", matchfile = comp_match)comp <- spillover(matched_fs, fsc ="FSC-H", ssc = "SSC-H", prematched = TRUE)
3. 圈门以及可视化
首先我们看下通过算法的自动圈门过程。我们看下flowCore中的几种方法:

实例:
rectGate <-rectangleGate(filterId="Fluorescence Region", "FL1-H"=c(0,12), "FL2-H"=c(0, 12))result = filter(fs[[1]],rectGate) ###获取此区域的数据summary(result)##Fluorescence Region+: 9811 of 10000events (98.11%)
当然多个数据集也是可以的:
result = filter(fs,rectGate) ###获取此区域的数据summary(result)

最后还能针对不同的样本获取不同的门:
rg2 <- rectangleGate(filterId="nonDebris","FSC-H"=c(300,Inf))rg3 <-rectangleGate(filterId="nonDebris","FSC-H"=c(400,Inf))flist <- list(rectGate, rg2, rg3)names(flist) <- sampleNames(foo)fr3 <- filter(foo, flist)
接下来我们看下在flowStats是如何实现这个过程的,其中主要的函数是norm2Filter,我们直接看下实例:
library(flowStats)morphGate <- norm2Filter("FSC-H","SSC-H", filterId="MorphologyGate", scale=2)smaller <- Subset(fs, morphGate)
接下来我们看下如何进一步对门数据进行k-mean聚类分析:
split(smaller[[1]],kmeansFilter("FSC-H"=c("Low","Medium","High"),filterId="myKMeans"))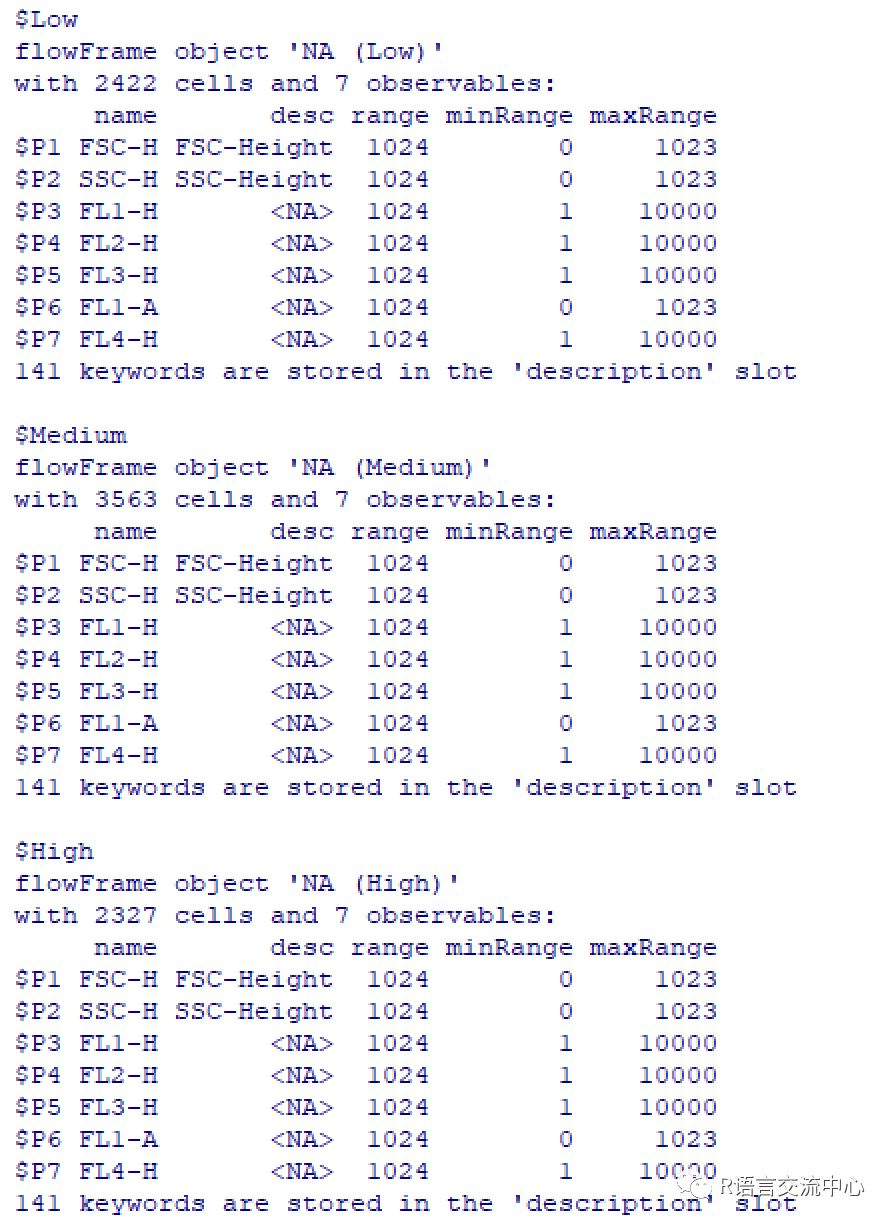
当然也可以直接对所有的样本进行直接的分组:
split(smaller,kmeansFilter("FSC-H"=c("Low", "Medium","High"), filterId="myKMeans"))那么如何自动获取多个门的数据并进行可视化,需要用到下面的函数,我们直接用一个实例来说明:
fcsfiles <- list.files(pattern ="CytoTrol", system.file("extdata", package ="flowWorkspaceData"), full = TRUE)fs <- read.flowSet(fcsfiles)gs <- GatingSet(fs)#构造门数据集fs_comp <- getData(gs)autoplot(fs_comp[[1]], "B710-A")+ ggtitle("raw")
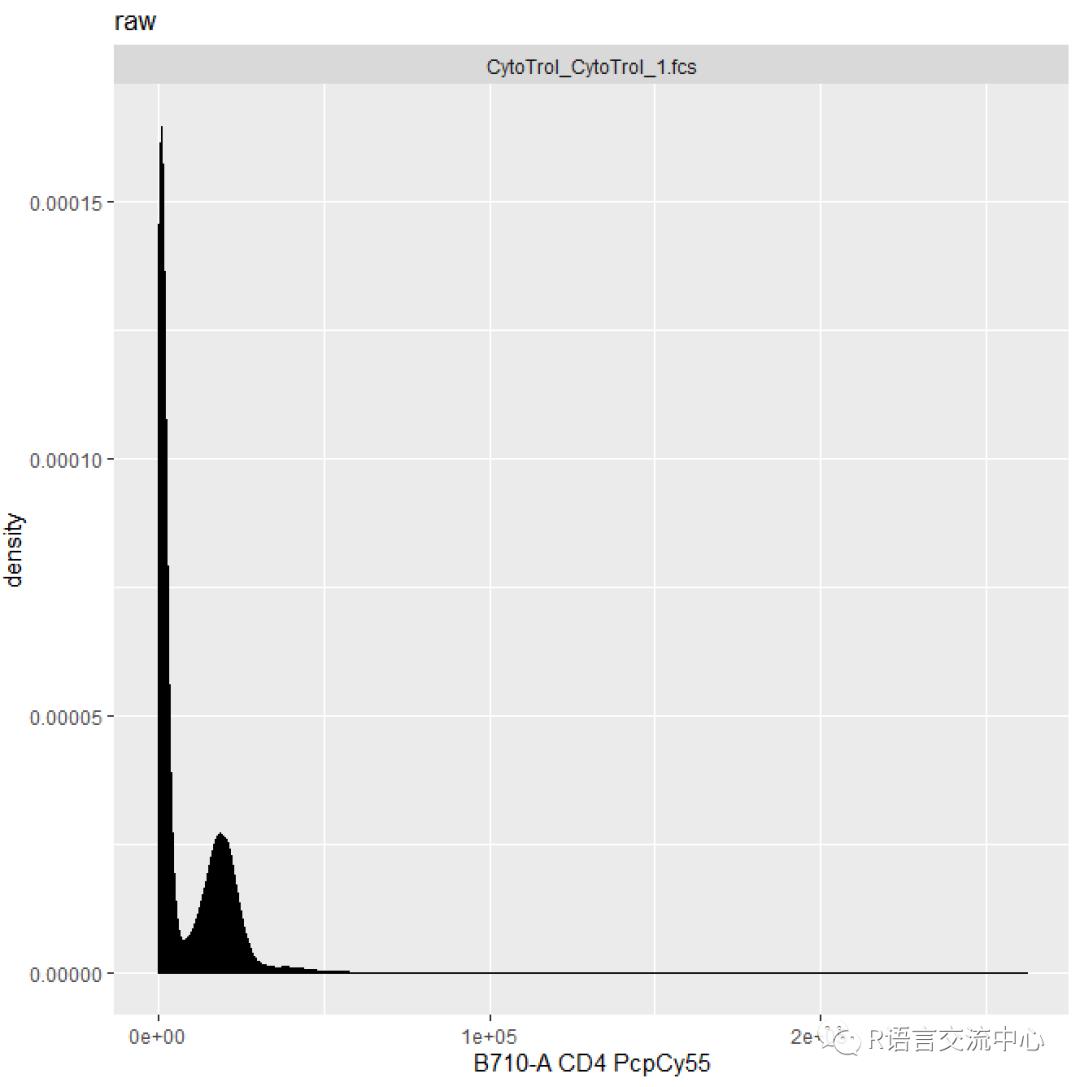
我们看到参数有-A和-H等具体是什么意思。其实早有定义A是area的缩写,H是height的缩写,W是width的缩写。那么有时候需要增加一个门的话,其实此包还增加了这一部分功能:
rg1 <-rectangleGate("FSC-A"=c(50000, Inf), filterId="NonDebris")gs_pop_add(gs, rg1, parent ="root")gs_get_pop_paths(gs) ##获取当下的节点recompute(gs)autoplot(gs, "NonDebris")
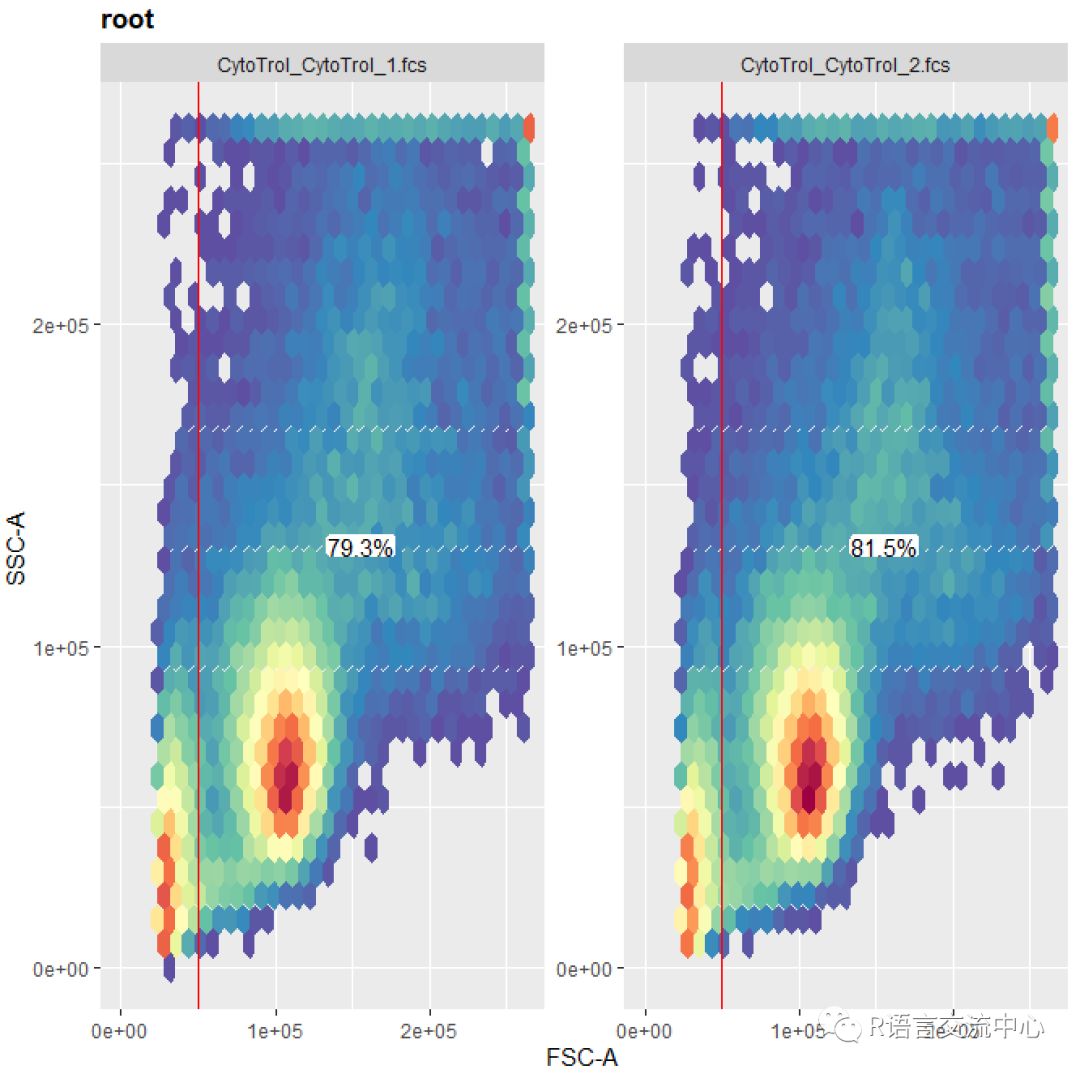
ggcyto(gs, aes(x = `FSC-A`)) +geom_density() + geom_gate("NonDebris")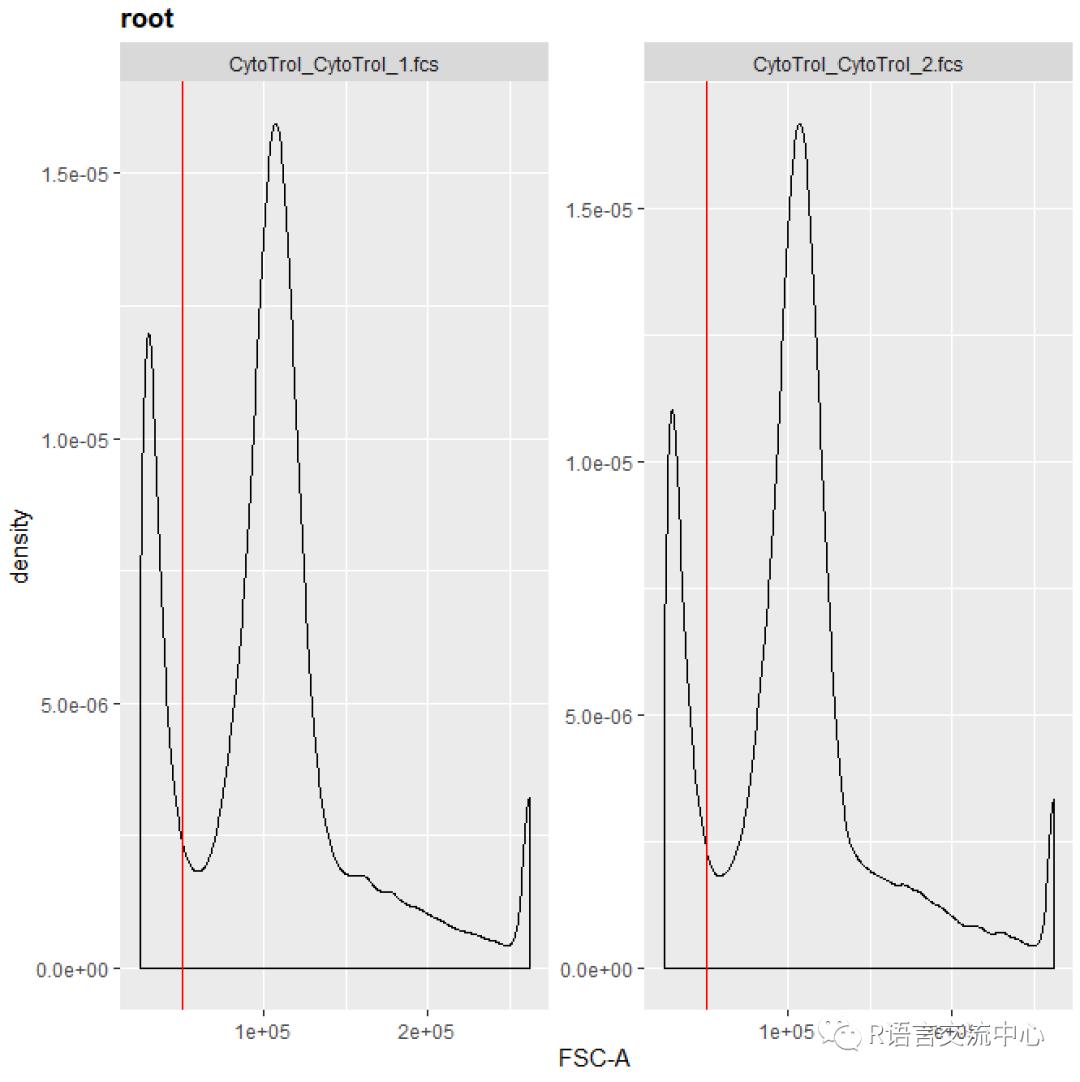
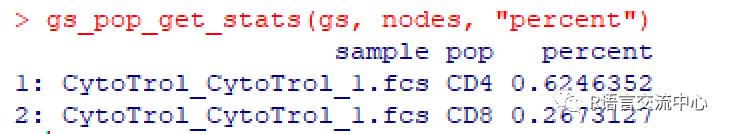
那如何获取每一部分的百分比呢,我们可以直接看下实例:
dataDir <-system.file("extdata",package="flowWorkspaceData")suppressMessages(gs <-load_gs(list.files(dataDir, pattern = "gs_manual",full = TRUE)))dt <- gs_pop_get_stats(gs)nodes <- c("CD4","CD8")gs_pop_get_stats(gs, nodes,"percent")
plot(gs)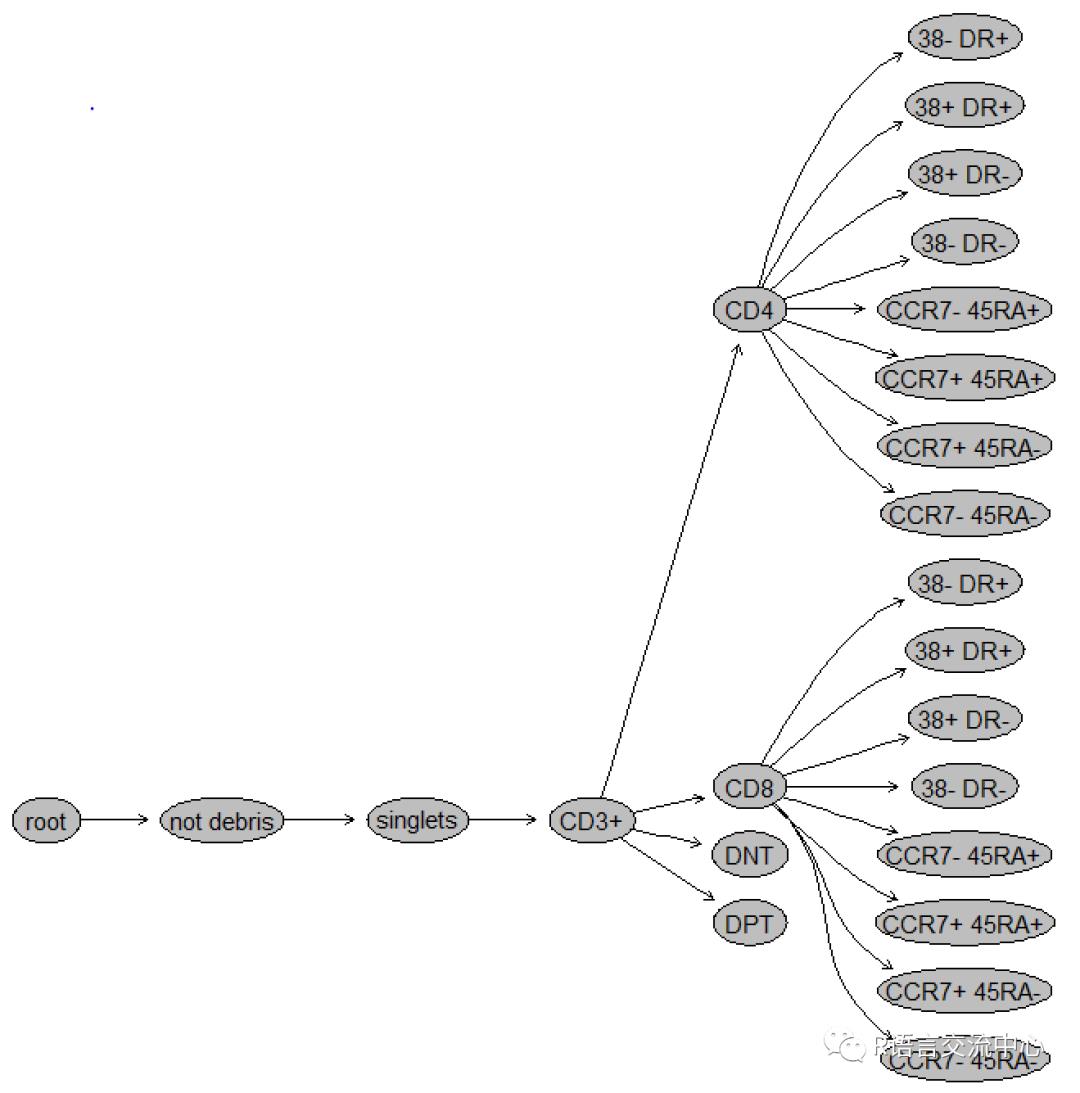
autoplot(gs[[1]])#可视化所有的门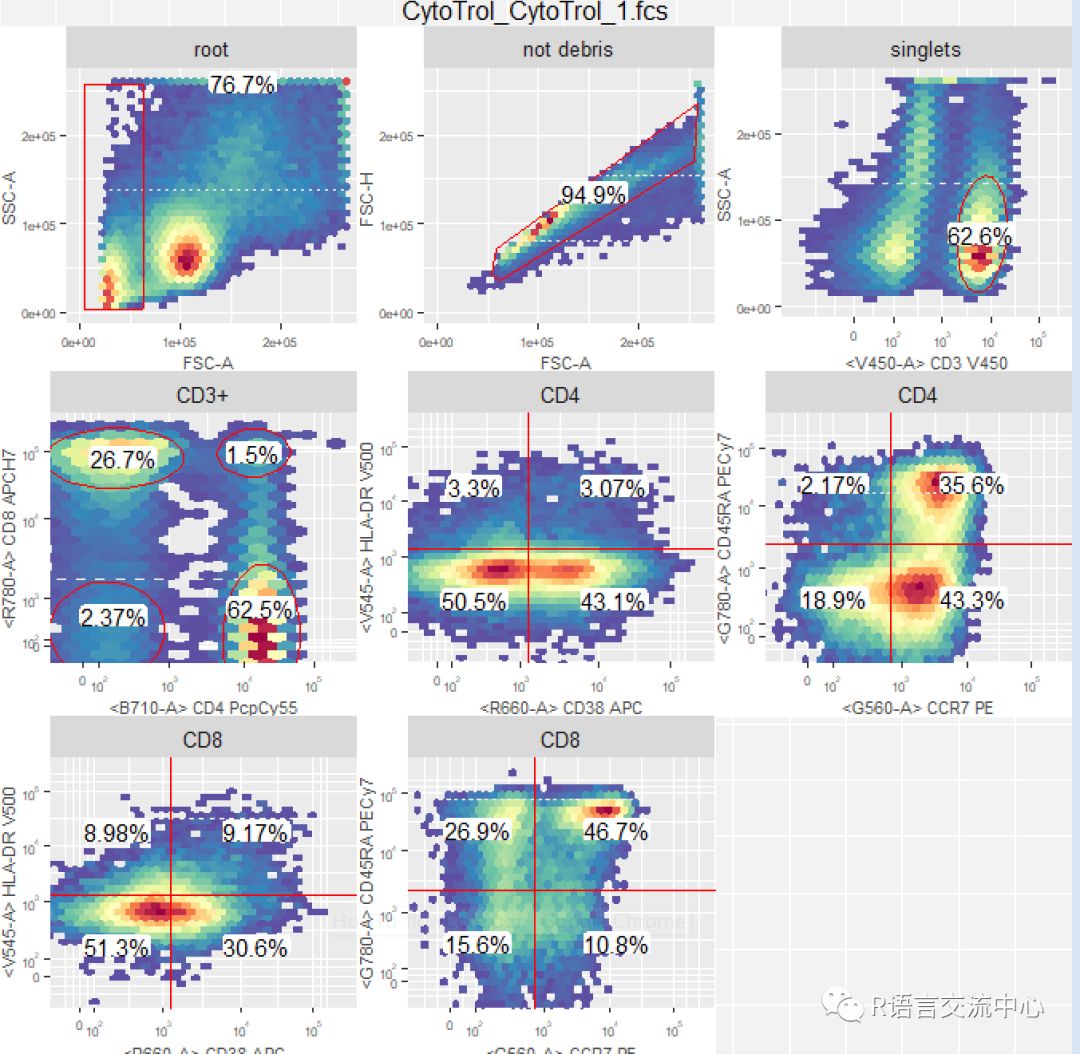
当然还可以更加细化门的分类,以及门的构成,在此我们就不再深究了。还有两个专门做自动圈门的包,我们就不细化讲解了当然如果我们想让可视乎更加的漂亮,也可以利用一些降维的方法操作原始数据,最终实现圈门。
欢迎大家学习交流!
以上是关于R语言实现流式细胞数据分析的主要内容,如果未能解决你的问题,请参考以下文章
流式仪器:福流生物-Flow NanoAnalyzer纳米流式检测仪