如何用R语言实现倾向评分匹配(Propensity Score Matching)
Posted 放化疗识鉴和疼痛管理
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了如何用R语言实现倾向评分匹配(Propensity Score Matching)相关的知识,希望对你有一定的参考价值。

陈按:倾向评分匹配可以用R语言实现,一般使用MatchIt包实现,这两天学习发现还可以使用nonrandom包实现,所以整理此贴予以梳理,亲自进行验证代码可以使用,就是包安装过程中会出现一些问题,故把整个过程以及代码演示在帖子中,供大家参考。
倾向评分匹配(Propensity Score Matching,简称PSM)是一种统计学方法,用于处理观察研究(Observational Study)的数据。在观察研究中,由于种种原因,数据偏差(bias)和混杂变量(confounding variable)较多,倾向评分匹配的方法正是为了减少这些偏差和混杂变量的影响,以便对实验组和对照组进行更合理的比较。这种方法最早由Paul Rosenbaum和Donald Rubin在1983年提出,一般常用于医学、公共卫生、经济学等领域。
PSM的步骤
计算倾向值(采用logistic回归)进行得分匹配
得分匹配的几种方法:
(1)最邻近匹配(Nearest neighbor matching, NNM)(是否使用卡尺 with or without caliper)以倾向得分为依据,在控制组样本中向前或向后寻找最接近干预组样本得分的对象,并形成配对。
(2)半径匹配(Radius matching)设定一个常数r(可理解为区间或范围,一般设定为小于倾向得分标准差的四分之一),将实验组中得分值与控制组得分值的差异在r内的进行配对。
(3)核匹配(Kernel Matching)将干预组样本与由控制组所有样本计算出的一个估计效果进行配对,其中估计效果由实验组个体得分值与控制组所有样本得分值加权平均获得,而权数则由核函数计算得出。
评定匹配后的平衡性
计算平均干预效果(ATT)
进行敏感性分析
适用情形
倾向评分匹配法适用于两类情形。第一,在观察研究中,对照组与实验组中可直接比较的个体数量很少。在这种情形下,实验组和对照组的交集很小,比如治疗组健康状况最好的10%人群与非治疗组健康状况最差的10%人群是相似的,如果将这两个重合的子集进行比较,就会得出非常偏倚的结论。第二,由于衡量个体特征的参数很多,所以想从对照组中选出一个跟实验组在各项参数上都相同或相近的子集作对比变得非常困难。在一般的匹配方法中,我们只需要控制一两个变量(如年龄、性别等)即可,就可以很容易从对照组中选出一个拥有相同特征的子集,以便与实验组进行对比。但是在某型情形下,衡量个体特征的变量会非常多,这时想选出一个理想的子集变得非常困难。经常出现的情形是,控制了某些变量,但是在其他变量上差异很大,以至于无法将实验组和对照组进行比较。倾向评分匹配通过使用逻辑回归模型来决定评分。
在使用nonrandom包来进行倾向得分匹配的话,需要先安装下面两个R包:
install.packages("nonrandom")
install.packages("tableone")
直接使用上面命令,可能就难以安装成功,会出现警告:
package ‘nonrandom’ is not available (for R version 3.6.3)
从github上下载zip包后离线手动安装
install.packages("devtools")
install.packages("rJava")
library(rJava)
library(devtools)
library(usethis)
devtools::install_local("C://Users//TD//Desktop//nonrandom-master.zip")
当然也可以不离线手动下载,直接网上下载:
install.packages("devtools")
install.packages("rJava")
library(rJava)
library(devtools)
library(usethis)
library(rJava)###java环境变量配置
devtools::install_github("cran/nonrandom")
如果下面的library命令能加载执行,则说明包安装成功。
library(nonrandom)
library(tableone)
接下来我把网络上其他人所实践的案例,根据他提供的R代码操作了一遍,是切实可行的,所以我整理在这里,供大家参考。自己知道如何根据自己的数据进行代码修改就行。
library(tableone)
library(nonrandom)
mydata1 <- read.csv("mydata1.csv")
#载入数据,包含422条记录,8个变量
View(mydata1)
结果:
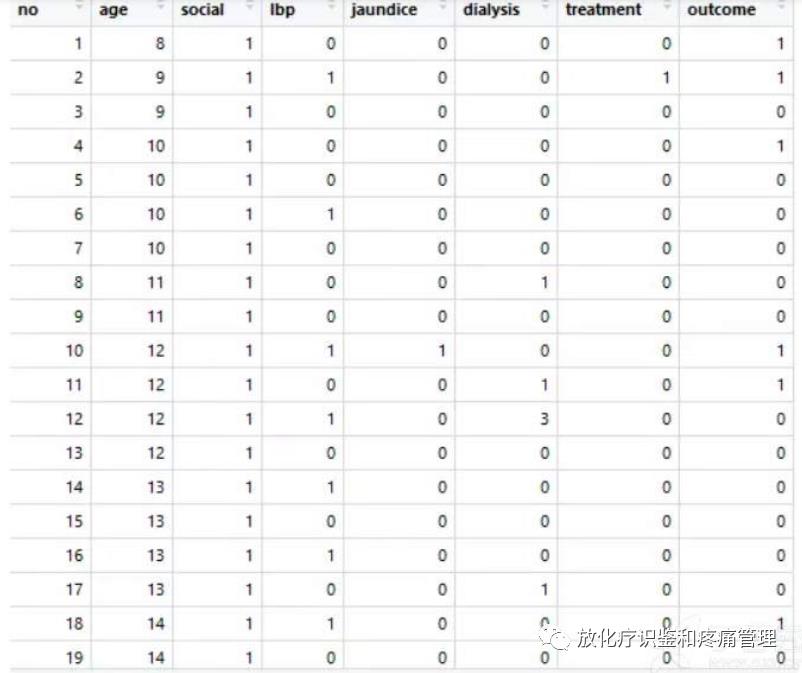
输入:
stable <- CreateTableOne(vars=c("age","social","lbp","jaundice","dialysis"),strata="treatment",data=mydata1,factorVars=c("social","lbp","jaundice","dialysis"))
print(stable,showAllLevels = TRUE)
结果:
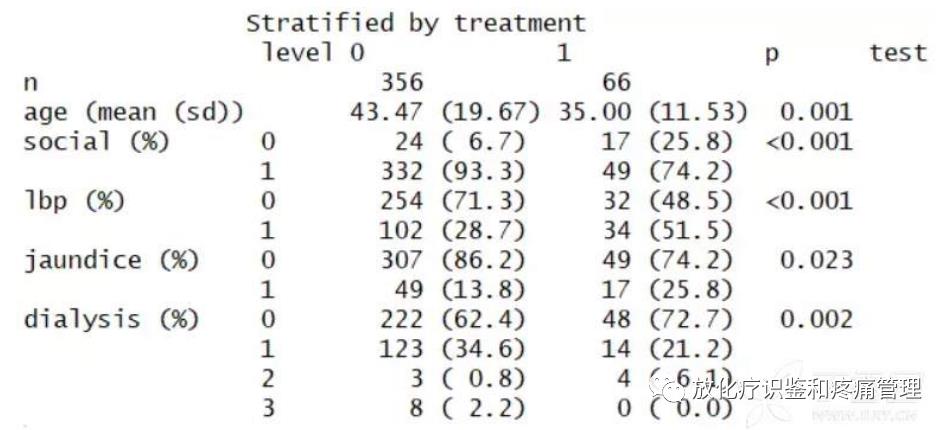
#显示原始数据treatment分层下各协变量不均衡。
输入:
mydata.ps <- pscore(data= mydata1, formula =
treatment~age+social+lbp+jaundice+dialysis)
plot.pscore(mydata.ps,with.legend=T,legend.cex=1,
main = "PS distribution",
par.1=list(col="red",lwd=2),
par.0=list(col="blue",lwd=2,lty=2),
xlim=c(-0.5,1))
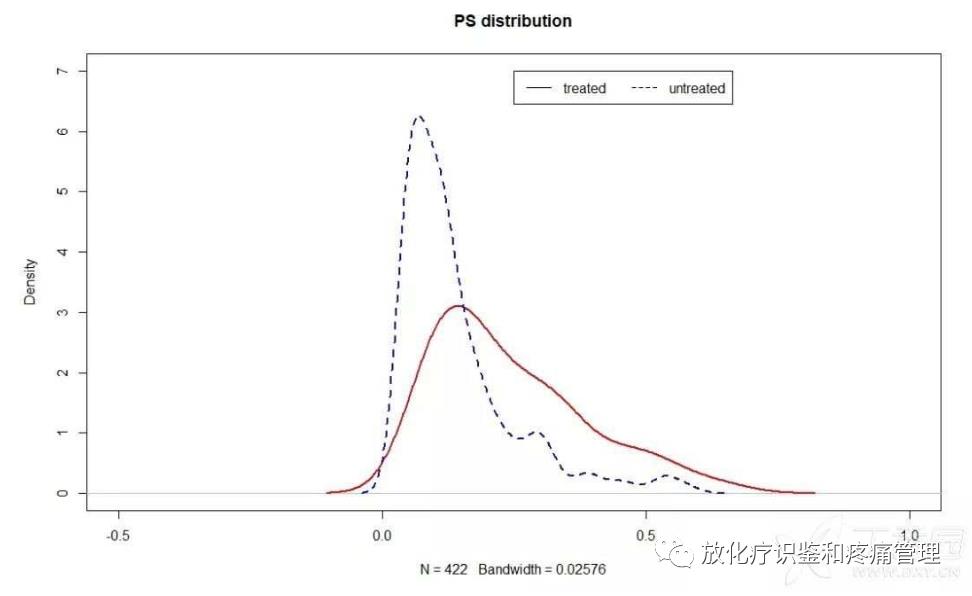
结果:#显示处理组和非处理倾向评分密度分布,par.1处理组作图参数,#par.0非处理组作图参数。
输入:
mydata.ps$data #显示倾向评分数据
summary(mydata.ps) #显示logistic回归模型
结果:(略)
输入:
mydata.match <- ps.match(object = mydata.ps,
who.treated =1,
ratio = 1,
caliper = "logit",
x =0.2,
givenTmatchingC = T,
matched.by = "pscore",
setseed = 12345)
#利用ps.match函数进行倾向评分匹配,who.treated =1,表示1代表处理组,ratio确定匹配比例,x代表得分容差,givenTmatchingC = T表示用未处理记录去匹配处理组记录。
输入:summary(mydata.match)
结果:#成功匹配66条记录。
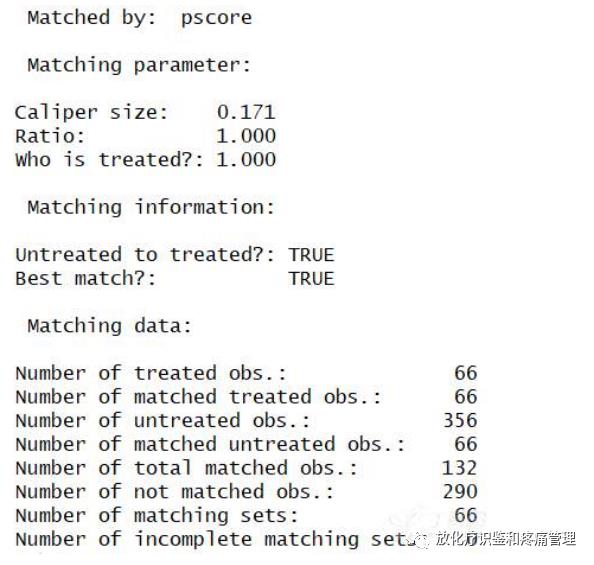
输入:pair<- mydata.match$data.matched #生成匹配后的数据
输入:dim(pair)
结果:
[1] 132 10 #132条记录,10个变量
输入:
stable1 <- CreateTableOne(vars=c("age","social","lbp","jaundice","dialysis"),
strata="treatment", data=pair,factorVars=c("social","lbp","jaundice","dialysis"))
print(stable1,showAllLevels = TRUE)
#对匹配后的数据进行统计分析
结果:
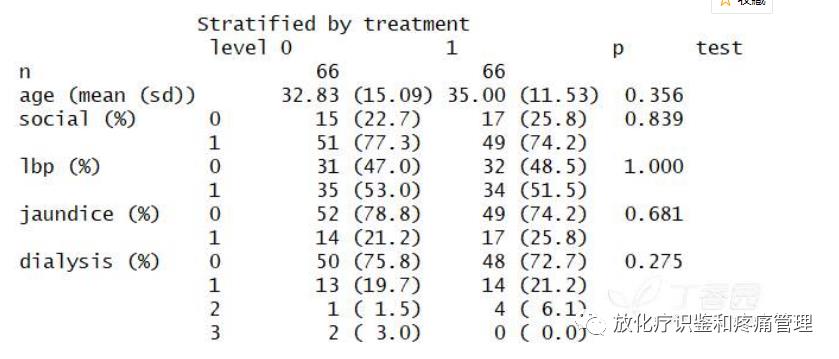
#显示各协变量差异无统计学意义,匹配效果较理想。
输入:
dist.plot(object=mydata.match,
sel=c("social"),
compare=T,
lable.match=c("original data","matched sample"))
#输出分类变量匹配前后的直条图,可更换其它分类变量以显示结果。
结果:
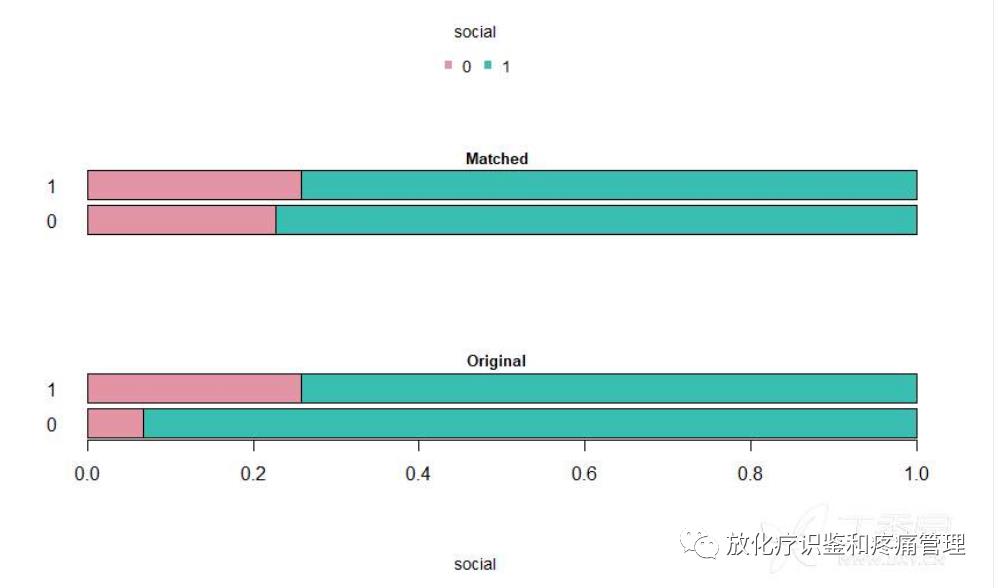
输入:
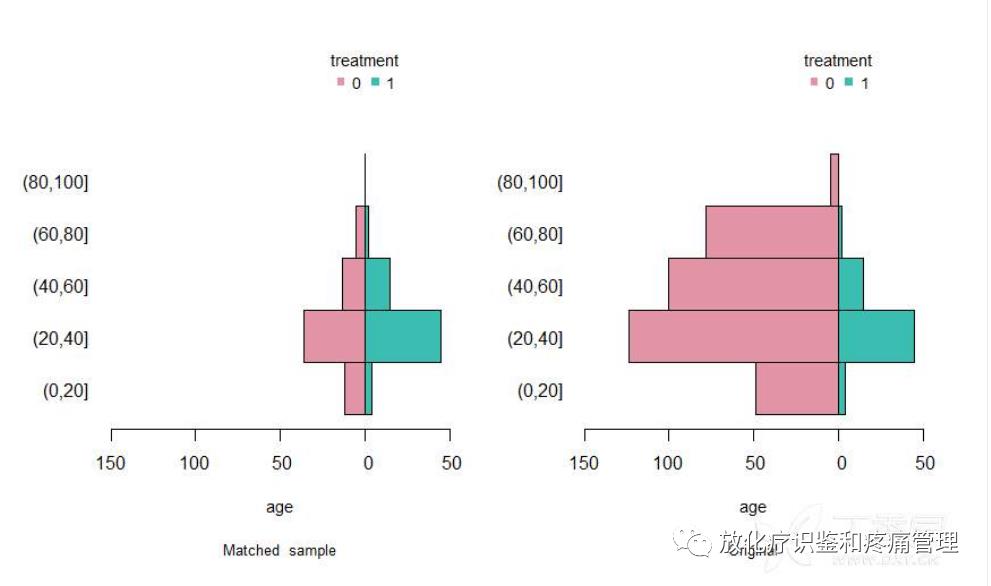
dist.plot(object=mydata.match,
sel=c("age"),
compare=T,
plot.type=2,
lable.match=c("original data","matched sample"))
#输出定量变量匹配前后的频数分布图
结果:
在这里补充一下:会发现上面2张图片显示不全或者不能显示,如何解决RGui和RStudio中"plot.new() : figure margins too large"问题呢,其实很简单,就是把RGui或者RStudio的窗口拖大,即可得到解决。
输入:
diffm <- ps.balance(object=mydata.match,
sel=c("age","social","lbp","jaundice","dialysis"),
cat.levels = 2,
method="classical",
alpha=5)
diffm
#对匹配后的数据进行均衡性假设检验, cat.levels = 2,表示数值大于2的变量的定量变量,method="classical",默认对定量和分类变量进行ttest和chisq.test,alpha=5设定显著性水平为0.05。
结果:

#此结果与前面stable1 的结果一致。
输入:
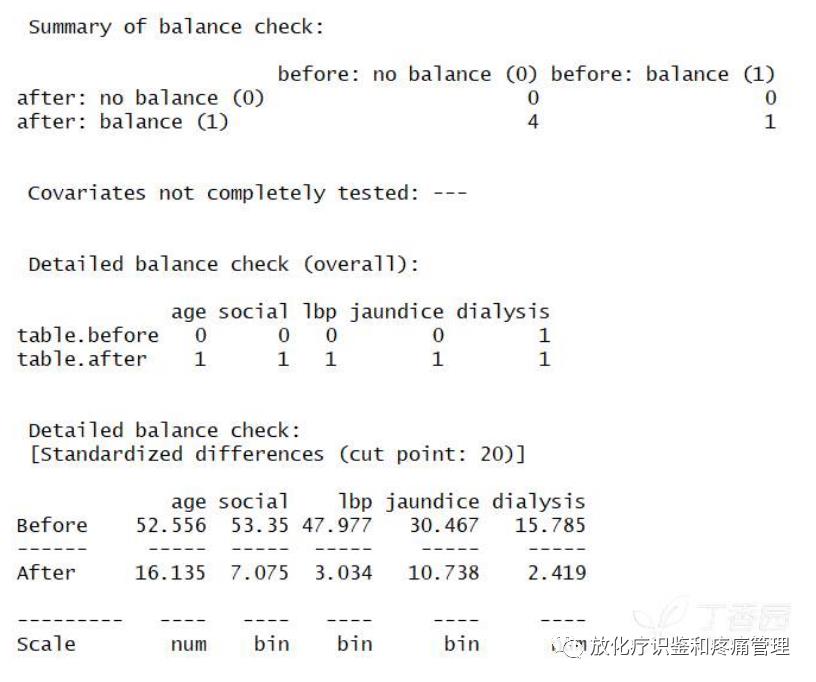
diffm1 <- ps.balance(object=mydata.match,
sel=c("age","social","lbp","jaundice","dialysis"),
cat.levels = 2,
method="stand.diff",
alpha=20)
diffm1
# method="stand.diff",表示采用标准差异法进行均衡性检验, alpha=20设定差异的临界值为0.2。
结果:
输入:
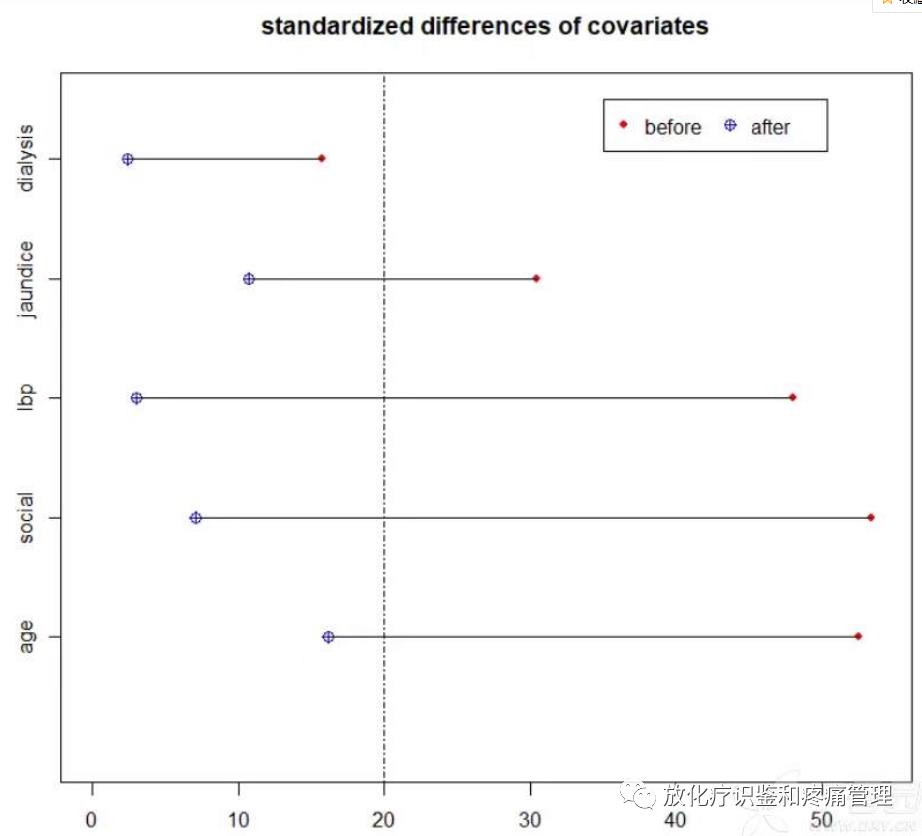
plot.stdf(x = diffm1,sel = c("age","social","lbp","jaundice","dialysis"),
main = "standardized differences of covariates",
pch.p=c(20,10),col.p=c("red","blue"),
legend.cex=1,legend.xy=c(35,6.5))
#图示化匹配前后协变量标准化差异值。
结果:
整个过程就是这样了,关键还在于多实践。
---------------------------------(end)---------------------------------
免责声明:本公众平台纯粹为了学术和公益需要,不牵涉任何经济商业利益,方便自己不断的学习和提升临床技能,加强科研锻炼,也同时方便患者朋友在线答疑。如果前面有任何帖子牵涉到专家版权等,请与我联系,我将在第一时间予以删除,谢谢!
欢迎大家关注,每一次真实案例的记录,是帮助自己总结诊治思路,更好的学习成长,用自己的仁心仁术让患者受益!
我们宁波大学附属人民医院(原鄞州医院)放化疗中心是宁波市肿瘤学重点扶植学科,技术力量雄厚,目前拥有宁波市最齐全的放射治疗仪器设备,也是放化疗一体的规范化治疗中心。我们放化疗中心总共有4个病区,基本上都是硕士及以上学历,人才梯队建设完善,我们将竭诚以优质的技术水准为您服务。
有既往就诊资料或者影像资料,最好能亲自前往我科室咨询,我的科室目前在宁波大学附属人民医院9号楼放化疗中心四楼(34病区)。
公交线路一: 3、15、16、103、216、302、342、354、357、506、517、620到宁波大学附属人民医院、银殿宾馆站下。
公交线路二: 3、101、103、105、117、342、367、514、517、527、560、804、901到甬港新村下。
公交线路三: 10、101、111、105、108、360、367、506、514、560、514、560、804 到舟孟新村下。
地铁①号线: 舟孟北路站B口
公共自行车: 宁波大学附属人民医院(南门)15桩、东城百汇20桩、宁波大学附属人民医院东25桩。
以上是关于如何用R语言实现倾向评分匹配(Propensity Score Matching)的主要内容,如果未能解决你的问题,请参考以下文章