R语言 | 加权基因共表达网络分析(WGCNA)
Posted 生物空间站
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言 | 加权基因共表达网络分析(WGCNA)相关的知识,希望对你有一定的参考价值。

示例数据、R代码的百度盘链接(提取码,zb33):
https://pan.baidu.com/s/1UE8c5ev-gCA-48_6vCcUKQ
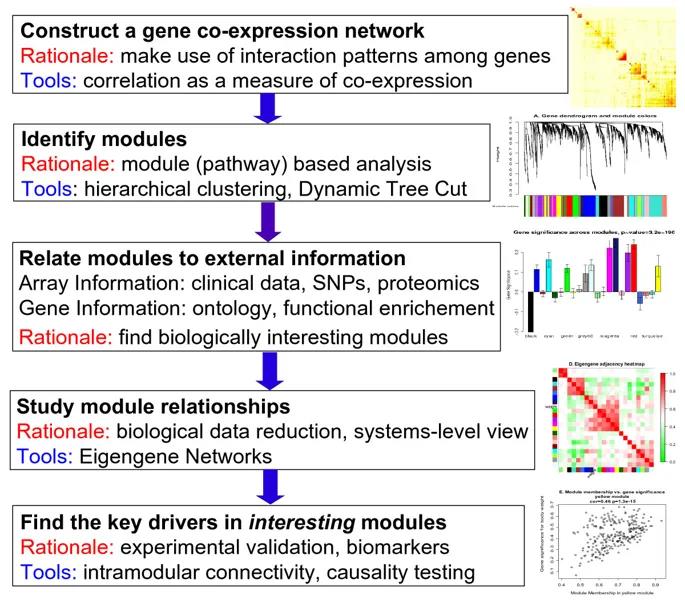
WGCNA的加权原理
在下文介绍具体过程之前,首先来理解一下WGCNA的加权原理。
传统方法上,描述两个基因间的关联程度,可通过计算表达值间的获得。为了构建关联网络,通常指定一个筛选阈值,如相关系数大于0.8以上,作为两个基因间具有强关联程度的依据。随后在网络中,节点表示基因,若两个基因间的相关系数大于指定的阈值,则会以边连接以表示两者间可能存在相互作用。
但是基于固定阈值法的缺点在于,阈值是人为定义的,将会忽略很多潜在关联。例如,0.79就是不相关吗?同时,这种一刀切的方法也会丢失基因的变化趋势信息,将难以在网络中描述相关性的强弱关系。
为了解决这些问题,提出了“加权”的思想。WGCNA的做法是对基因表达值之间的相关系数取β次幂,直接结果是把基因间相关性的强弱的差异放大。这样做的好处是使强弱关系更为分明,有利于后续聚类(模块)识别。
如下所示,对于基因i和j,相关系数为rij,取β次幂后获得aij,最终表征基因的相关强度。
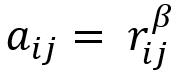
因此,最终网络中基因间的相关强度,取决于β次幂的选择,其取值的高低直接影响模块的构建和模块内基因的划分。β值根据接近无标度网络(scale-free network)的最低值确定,下文的计算过程中,power值的选择即为确定β值的过程。
根据这个思想,计算所有基因间表达水平的关联强度,获得邻接矩阵。并进一步,计算拓扑重叠矩阵(TOM),简单来说如果基因i和j有很多相同的邻接基因,那么TOMi,j就高,意味着两基因间有相似的表达模式。因此,TOM矩阵用于反映基因间表达的相似性。
在这些过程中,不再根据某关联强度阈值或者相似度阈值做筛选,因此不会丢失基因的变化趋势信息。过程中综合考虑所有给定基因间的表达关系,根据基因间表达的相似性,进行层次聚类划分功能模块,进而识别模块特征基因、确定功能模块中基因表达和性状的关联等,详见下文过程。
输入数据示例
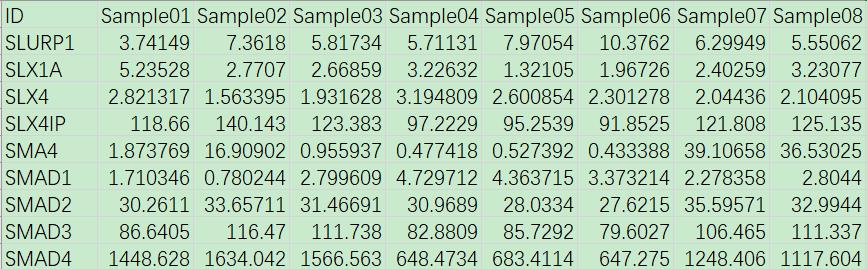
网盘附件“gene_express.txt”为某项关于肿瘤的研究中通过转录组测序获得的患者肿瘤组织的基因表达值矩阵,包含了24个样本中基因表达的FPKM值信息。
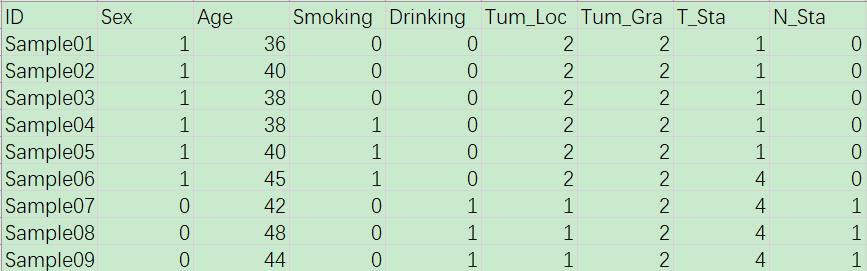
“trait.txt”则记录了24个肿瘤组织样本所来源的患者的临床资料信息,包括性别、年龄、是否有吸烟史、饮酒史以及肿瘤分期、生存状态等信息。
接下来,通过WGCNA识别肿瘤组织中基因的共表达概况,并期望寻找与疾病进展相关的基因集。
加权网络构建及共表达模块划分
首先加载R包,并读取基因表达值矩阵。
可以进行一些前处理,比方说处理缺失值,按表达水平过滤低表达的基因等,避免它们的影响,同时减少后续运算资源消耗。
#WGCNA 包通过 bioconductor 安装
#install.packages('BiocManager')
#BiocManager::install('WGCNA')
library(WGCNA)
#示例数据,基因表达值矩阵
gene <- read.delim('gene_express.txt', row.names = 1, check.names = FALSE)
#例如过滤低表达值的基因,只保留平均表达值在 1 以上的
gene <- subset(gene, rowSums(gene)/ncol(gene) >= 1)
#转置矩阵,行是基因,列是样本
gene <- t(gene)
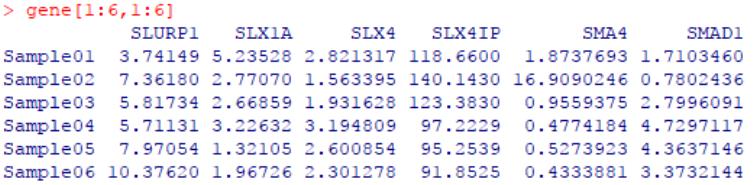
无标度拓扑分析确定最佳β值
为了构建加权共表达网络,首先需要确定一个软阈powers值(soft thresholding powers),作为上文提到的相关系数的β次幂,以建立邻接矩阵并根据连通度使基因分布符合无尺度网络。
pickSoftThreshold()可通过将给定的基因表达矩阵按表达相似度计算加权网络,并分析指定powers值(构建一组向量,传递给powerVector)下网络的无标度拓扑拟合指数以及连通度等信息,目的是帮助为网络构建选择合适的powers值。
##power 值选择
powers <- 1:20
sft <- pickSoftThreshold(gene, powerVector = powers, verbose = 5)
#拟合指数与 power 值散点图
par(mfrow = c(1, 2))
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2], type = 'n',
xlab = 'Soft Threshold (power)', ylab = 'Scale Free Topology Model Fit,signed R^2',
main = paste('Scale independence'))
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels = powers, col = 'red');
abline(h = 0.90, col = 'red')
#平均连通性与 power 值散点图
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab = 'Soft Threshold (power)', ylab = 'Mean Connectivity', type = 'n',
main = paste('Mean connectivity'))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels = powers, col = 'red')
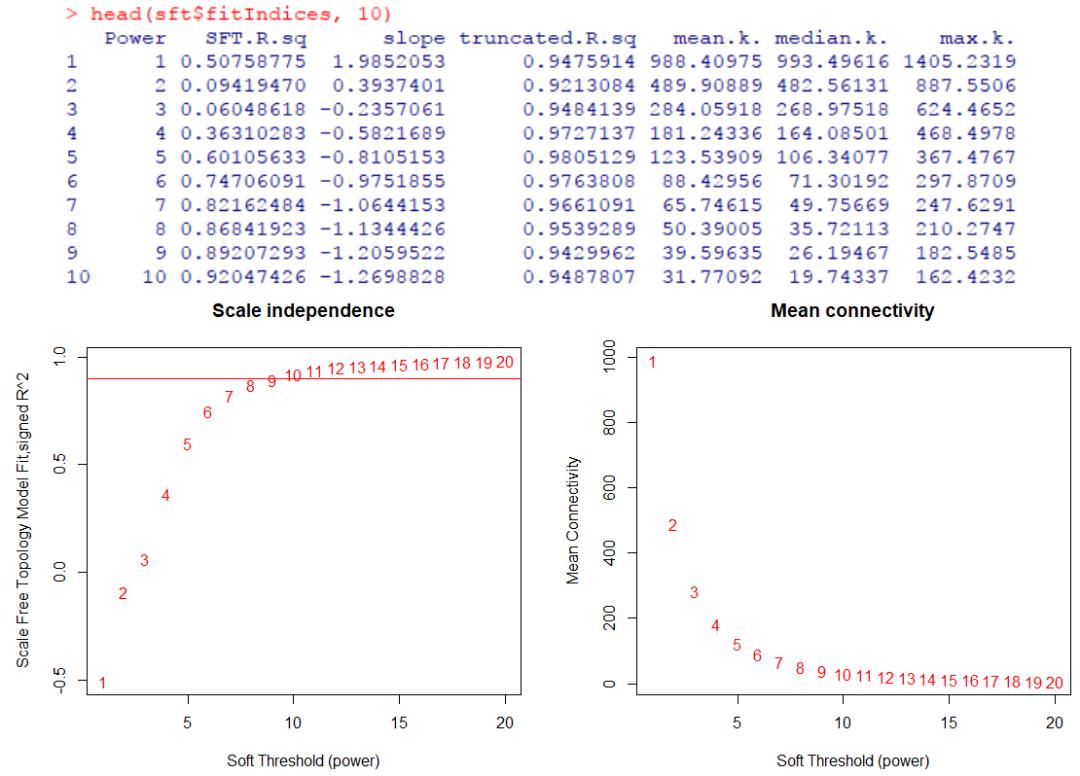
左图中纵轴展示了无标度拓扑拟合指数R2(scale free topology fitting index R2,即统计结果中的SFT.R.sq列数值),之所以有负值是因为又乘以了slope列数值的负方向,仅关注正值即可;右图纵轴展示了网络的平均连通度。
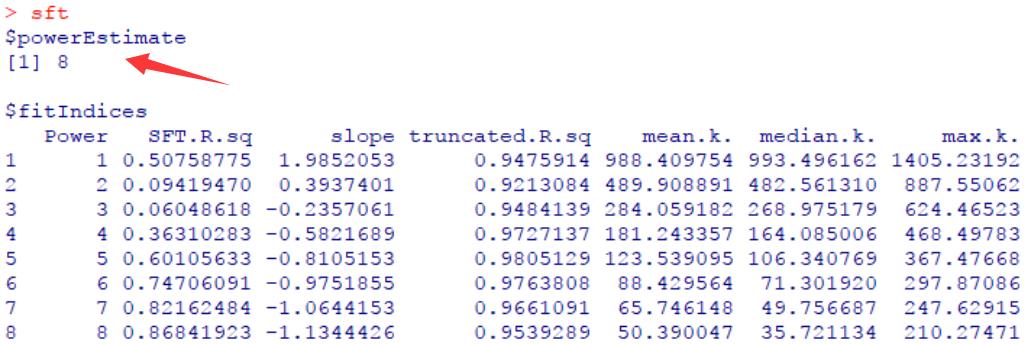
综合考虑这些指标选择合适的软阈powers值,使R2可能大但也要保证连通度不能太低。看到网上有教程中提到,一般选择R2大于0.85时的power值,那么在该示例中则需选择powers=8。
pickSoftThreshold()自身也估计了一个最佳的参考值,可以看到同样为powers=8。
获得基因共表达相似度矩阵
接下来,就使用上述估计的最合适的power值,构建无标度网络。
首先基于基因表达值矩阵计算基因间表达的相似度,获得邻接度矩阵,并进一步计算拓扑重叠矩阵(topological overlap matrix,TOM)。
#上一步估计的最佳 power 值
powers <- sft$powerEstimate
#获得 TOM 矩阵
adjacency <- adjacency(gene, power = powers)
tom_sim <- TOMsimilarity(adjacency)
rownames(tom_sim) <- rownames(adjacency)
colnames(tom_sim) <- colnames(adjacency)
tom_sim[1:6,1:6]
#输出 TOM 矩阵
#write.table(tom_sim, 'TOMsimilarity.txt', sep = ' ', col.names = NA, quote = FALSE)
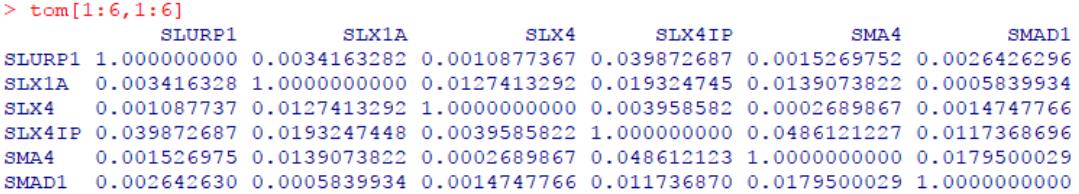
TOM矩阵中的值就反映了两两基因间共表达的相似度,取值0-1,基因间共表达相似度越高,值越趋于1。
共表达模块识别
上述获得了基因间的共表达相似度矩阵,记录了基因间表达的相似性。可以进一步据此计算距离并绘制聚类树,查看相似度的整体分布特征。
#TOM 相异度 = 1 – TOM 相似度
tom_dis <- 1 - tom_sim
#层次聚类树,使用中值的非权重成对组法的平均聚合聚类
geneTree <- hclust(as.dist(tom_dis), method = 'average')
plot(geneTree, xlab = '', sub = '', main = 'Gene clustering on TOM-based dissimilarity',
labels = FALSE, hang = 0.04)
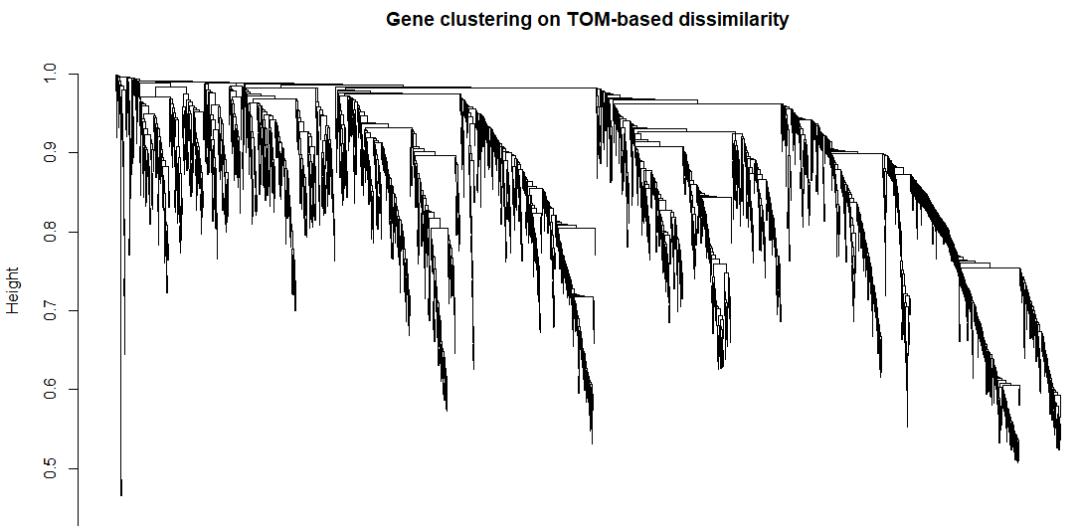
能够明显地看到,这些基因根据共表达相似性模式,可以划分为不同的聚类簇。每一簇中的基因间具有较高的共表达相似性,簇间的基因共表达相似性较低。
因此,就可以继续根据这种共表达模式(聚类簇特征),划分出几个更为明显的共表达模块。不难想到,同一模块内的基因具有较高的共表达相似性,即存在较高的协同,暗示它们参与了相似的调节途径,或者在相似的细胞区域中发挥功能。
#使用动态剪切树挖掘模块
minModuleSize <- 30 #模块基因数目
dynamicMods <- cutreeDynamic(dendro = geneTree, distM = tom_dis,
deepSplit = 2, pamRespectsDendro = FALSE, minClusterSize = minModuleSize)
table(dynamicMods)

这里共划分了19个模块,计数值为这些模块中对应的基因数量。
在文献中,这些模块通常通过“颜色”指代,例如我们经常会在文献中看到以“red module”、“lightgreen module”等作为模块的名称进行描述。
类似地,接下来为这些模块赋值颜色,并通过颜色名称命名不同的模块。
#模块颜色指代
dynamicColors <- labels2colors(dynamicMods)
table(dynamicColors)
plotDendroAndColors(geneTree, dynamicColors, 'Dynamic Tree Cut',
dendroLabels = FALSE, addGuide = TRUE, hang = 0.03, guideHang = 0.05,
main = 'Gene dendrogram and module colors')
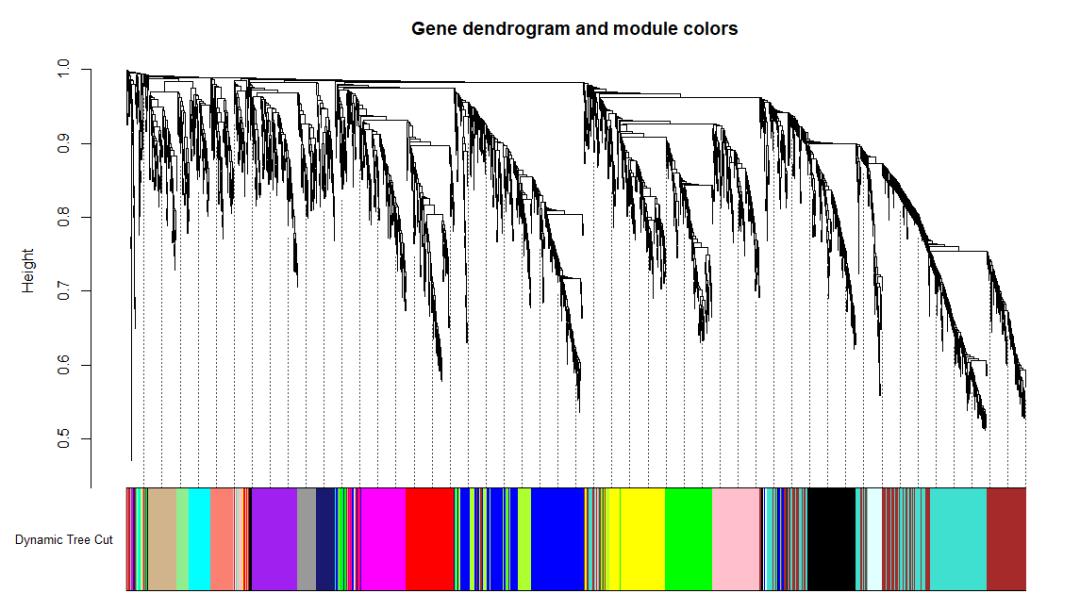
文献中也常见到这种表现形式,将基因共表达相似度矩阵绘制以热图,和表征模块的聚类树组合在一起展示。
#基因表达聚类树和共表达拓扑热图
plot_sim <- -(1-tom_sim)
#plot_sim <- log(tom_sim)
diag(plot_sim) <- NA
TOMplot(plot_sim, geneTree, dynamicColors,
main = 'Network heatmap plot, selected genes')
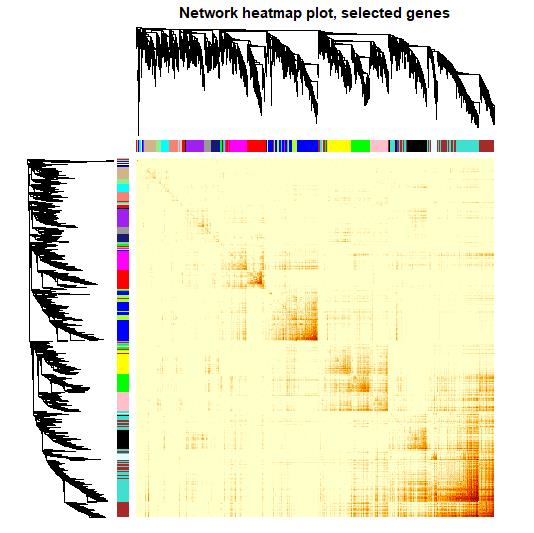
热图中,若基因间表达相似度越高,则颜色越深。可以看到,同一模块内的基因具有较高的共表达模式,而不同模块间则相差明显。
模块特征基因及其应用举例
所谓模块特征基因,定义为相应模块的表达矩阵的第一主成分,代表了该模块内基因表达的整体水平。
因此,模块特征基因并非某个具体的基因,而是一种“特征组合”。
模块特征基因计算
使用moduleEigengenes()可对基因表达值矩阵中的基因按模块划分后,执行特征分解,获取模块特征基因。
#计算基因表达矩阵中模块的特征基因(第一主成分)
MEList <- moduleEigengenes(gene, colors = dynamicColors)
MEs <- MEList$eigengenes
head(MEs)[1:6]
#输出模块特征基因矩阵
#write.table(MEs, 'moduleEigengenes.txt', sep = ' ', col.names = NA, quote = FALSE)
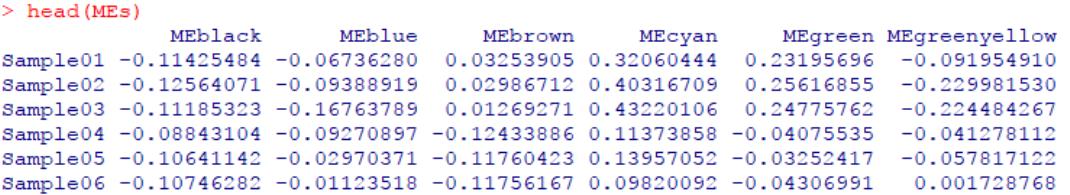
模块特征基因可用于反映各样本中,各基因共表达模块(具有相似的共表达模式)中基因的整体表达水平,或者使用它们替代整个模块中的基因执行特定的分析。
共表达模块的进一步聚类
例如对于上述划分的共表达模块,如果觉得模块过多,可以进一步合并,将相似度较高的模块聚合到一起。此时可以通过模块特征基因来实现,参考以下示例。
#通过模块特征基因计算模块间相关性,表征模块间相似度
ME_cor <- cor(MEs)
ME_cor[1:6,1:6]
#绘制聚类树观察
METree <- hclust(as.dist(1-ME_cor), method = 'average')
plot(METree, main = 'Clustering of module eigengenes', xlab = '', sub = '')
#探索性分析,观察模块间的相似性
#height 值可代表模块间的相异度,并确定一个合适的阈值作为剪切高度
#以便为低相异度(高相似度)的模块合并提供依据
abline(h = 0.2, col = 'blue')
abline(h = 0.25, col = 'red')
#模块特征基因聚类树热图
plotEigengeneNetworks(MEs, '', cex.lab = 0.8, xLabelsAngle= 90,
marDendro = c(0, 4, 1, 2), marHeatmap = c(3, 4, 1, 2))
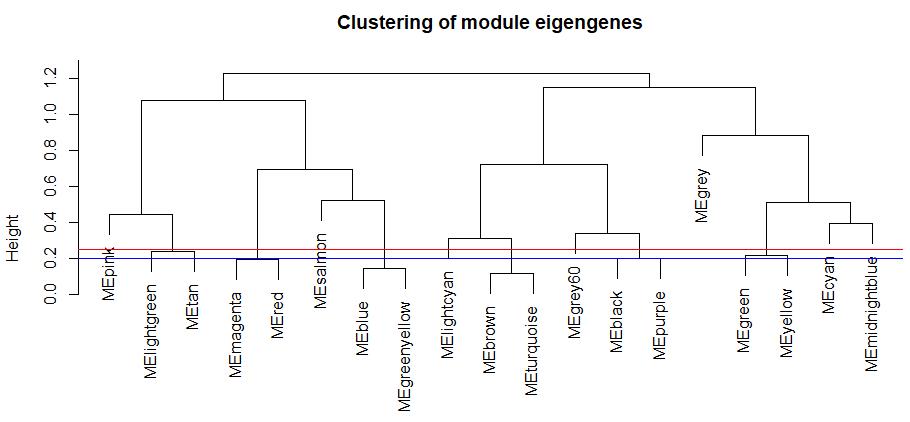
根据聚类树中所示模块间相似程度,定义一个合适的高度阈值,聚类树中低于该值的模块可以视为具有较高的相似程度,允许它们合并至一个簇中。
#相似模块合并,以 0.25 作为合并阈值(剪切高度),在此高度下的模块将合并
#近似理解为相关程度高于 0.75 的模块将合并到一起
merge_module <- mergeCloseModules(gene, dynamicColors, cutHeight = 0.25, verbose = 3)
mergedColors <- merge_module$colors
table(mergedColors)
#基因表达和模块聚类树
plotDendroAndColors(geneTree, cbind(dynamicColors, mergedColors), c('Dynamic Tree Cut', 'Merged dynamic'),
dendroLabels = FALSE, addGuide = TRUE, hang = 0.03, guideHang = 0.05)
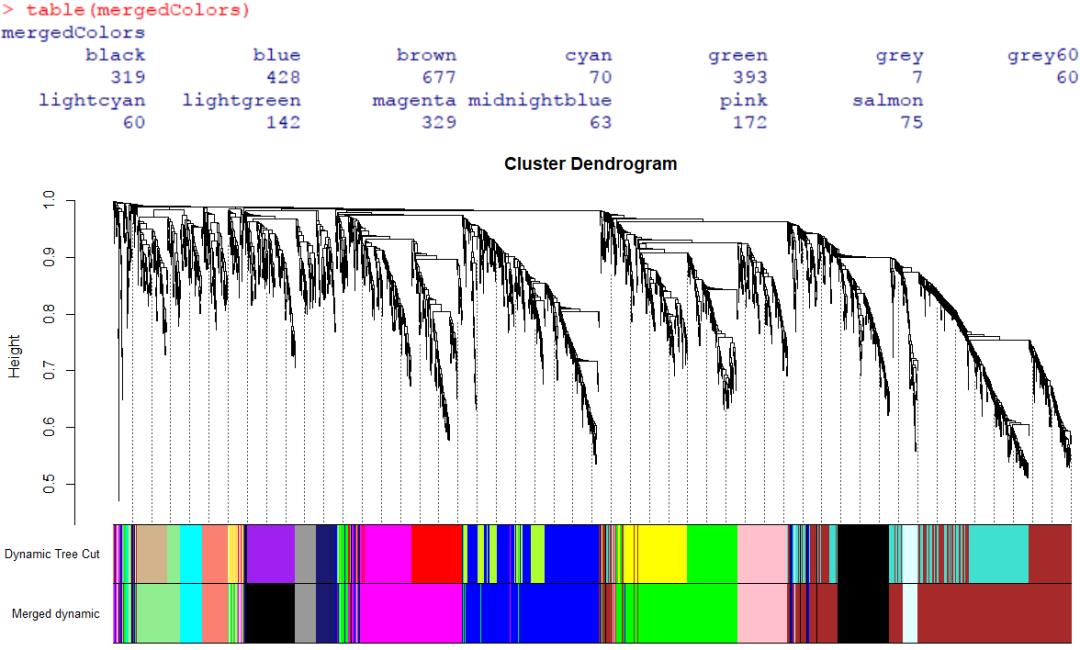
重新裁剪聚类树的结果中,保留了13个模块(之前是19个)。
共表达模块与与临床表型的关联分析
上文已经提到,同一模块内的基因具有较高的共表达相似性,即存在较高的协同,暗示它们参与了相似的调节途径,或者在相似的细胞区域中发挥功能。
怎样寻找具有生物学意义的共表达模块(基因集)呢?来看一个在文献中经常见到的例子。在肿瘤转录组分析中,在构建了WGCNA共表达模块后,通常将这些模块与患者的临床指标联系起来,以探索哪些基因间的协同作用可能与生理特征、疾病进展等密切相关。
由于共表达模块是一组基因集,而非单一的变量,因此无法直接计算其与临床表型资料的相关性。此时模块特征基因就可以派上用场。
#患者的临床表型数据
trait <- read.delim('trait.txt', row.names = 1, check.names = FALSE)
#使用上一步新组合的共表达模块的结果
module <- merge_module$newMEs
#患者基因共表达模块和临床表型的相关性分析
moduleTraitCor <- cor(module, trait, use = 'p')
moduleTraitCor[1:6,1:6] #相关矩阵
#相关系数的 p 值矩阵
moduleTraitPvalue <- corPvalueStudent(moduleTraitCor, nrow(module))
#输出相关系数矩阵或 p 值矩阵
#write.table(moduleTraitCor, 'moduleTraitCor.txt', sep = ' ', col.names = NA, quote = FALSE)
#write.table(moduleTraitPvalue, 'moduleTraitPvalue.txt', sep = ' ', col.names = NA, quote = FALSE)
#相关图绘制
textMatrix <- paste(signif(moduleTraitCor, 2), ' (', signif(moduleTraitPvalue, 1), ')', sep = '')
dim(textMatrix) <- dim(moduleTraitCor)
par(mar = c(5, 10, 3, 3))
labeledHeatmap(Matrix = moduleTraitCor, main = paste('Module-trait relationships'),
xLabels = names(trait), yLabels = names(module), ySymbols = names(module),
colorLabels = FALSE, colors = greenWhiteRed(50), cex.text = 0.7, zlim = c(-1,1),
textMatrix = textMatrix, setStdMargins = FALSE)
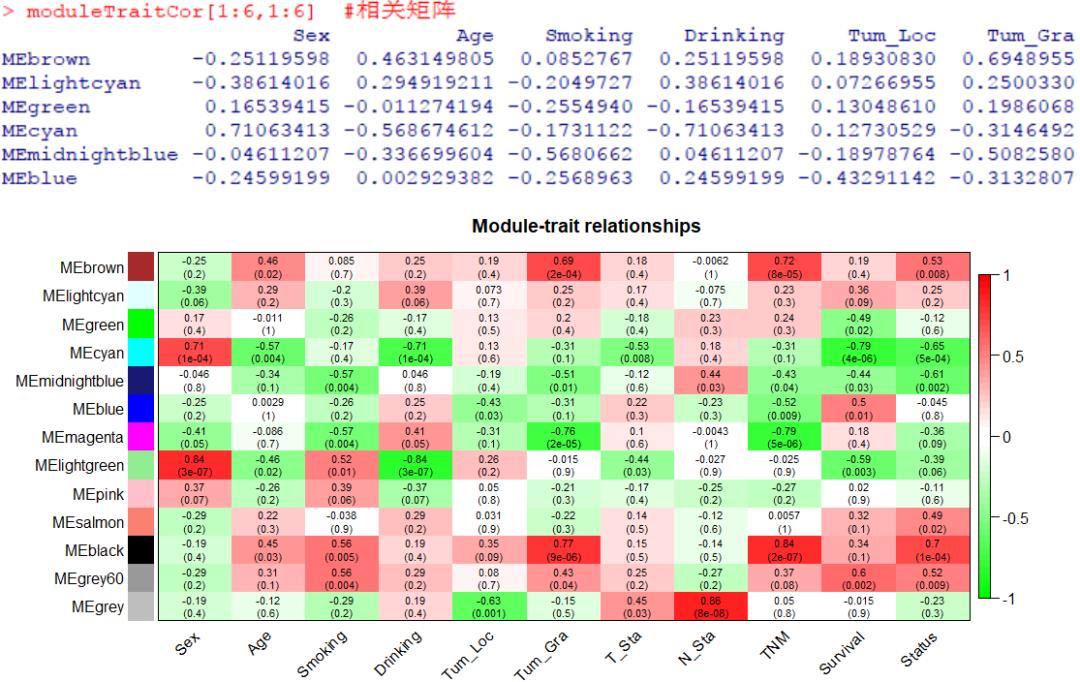
图中,每一行代表不同的基因共表达模块,每一列代表不同的临床表型,数值代表了相关系数,并分别通过红色和绿色区分正负相关,括号中的值为显著性p值。
由此可帮助评估,哪些模块中的基因可能与特定的生理特征、疾病进展等密切相关。
模块内基因及表达矩阵的提取
如果找到了感兴趣的共表达模块,最后就是将该模块所涉及的基因名称、基因的表达矩阵、或者基因间共表达相似度矩阵等提取出来,以便进行下游的分析,继续探索它们的功能。
例如,上述相关图中观察到“black”模块与肿瘤TNM分期存在非常显著的正相关,暗示该模块内的基因集参与了肿瘤进程。因此,期望获得与“black”模块有关的基因信息。
#基因与模块的对应关系列表
gene_module <- data.frame(gene_name = colnames(gene), module = mergedColors, stringsAsFactors = FALSE)
head(gene_module)
#“black”模块内的基因名称
gene_module_select <- subset(gene_module, module == 'black')$gene_name
#“black”模块内基因在各样本中的表达值矩阵(基因表达值矩阵的一个子集)
gene_select <- t(gene[,gene_module_select])
#“black”模块内基因的共表达相似度(在 TOM 矩阵中提取子集)
tom_select <- tom_sim[gene_module_select,gene_module_select]
#输出
#write.table(gene_select, 'gene_select.txt', sep = ' ', col.names = NA, quote = FALSE)
#write.table(tom_select, 'tom_select.txt', sep = ' ', col.names = NA, quote = FALSE)

例如后续可以根据基因名称,在STRING数据库中构建这些基因的蛋白互作网络,或者执行GO、KEGG功能富集分析或GSEA查看它们主要涉及到哪些通路的失调可能导致疾病发生和进展等,不再多说。
寻找模块中关键基因子集
已知“black”模块中的基因表达与肿瘤TNM分期存在非常显著的正相关,由于该模块中的基因数量非常多,因此期望进一步找出可能的驱动基因子集。
可通过计算各基因在“black”模块中的模块成员度(module membership),识别该模块的代表基因;同时将各基因的表达值与TNM分期表型作相关性分析,看哪些基因的高表达与TNM分期的进展显著相关。
#选择 black 模块内的基因
gene_black <- gene[ ,gene_module_select]
#基因的模块成员度(module membership)计算
#即各基因表达值与相应模块特征基因的相关性,其衡量了基因在全局网络中的位置
geneModuleMembership <- signedKME(gene_black, module['MEblack'], outputColumnName = 'MM')
MMPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneModuleMembership), nrow(module)))
#各基因表达值与临床表型的相关性分析
geneTraitSignificance <- as.data.frame(cor(gene_black, trait['TNM'], use = 'p'))
GSPvalue <- as.data.frame(corPvalueStudent(as.matrix(geneTraitSignificance), nrow(trait)))
#选择显著(p<0.01)、高 black 模块成员度(MM>=0.8),与 TNM 表型高度相关(r>=0.8)的基因
geneModuleMembership[geneModuleMembership<0.8 | MMPvalue>0.01] <- 0
geneTraitSignificance[geneTraitSignificance<0.8 | MMPvalue>0.01] <- 0
select <- cbind(geneModuleMembership, geneTraitSignificance)
select <- subset(select, geneModuleMembership>=0.8 & geneTraitSignificance>=0.8)
head(select)
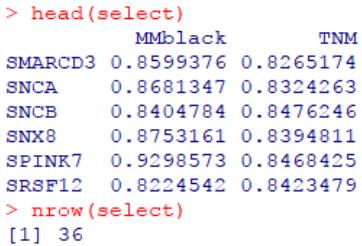
考虑多个条件后,识别了36个可能的候选标志物。
此外,还可结合差异表达分析的结果,例如肿瘤组织和正常组织相比哪些基因发生显著失调,并根据显著性、差异倍数变化程度等继续在这36个候选基因中进一步选择可能推动肿瘤进程的基因。
顺便作图观察这些基因的概况。
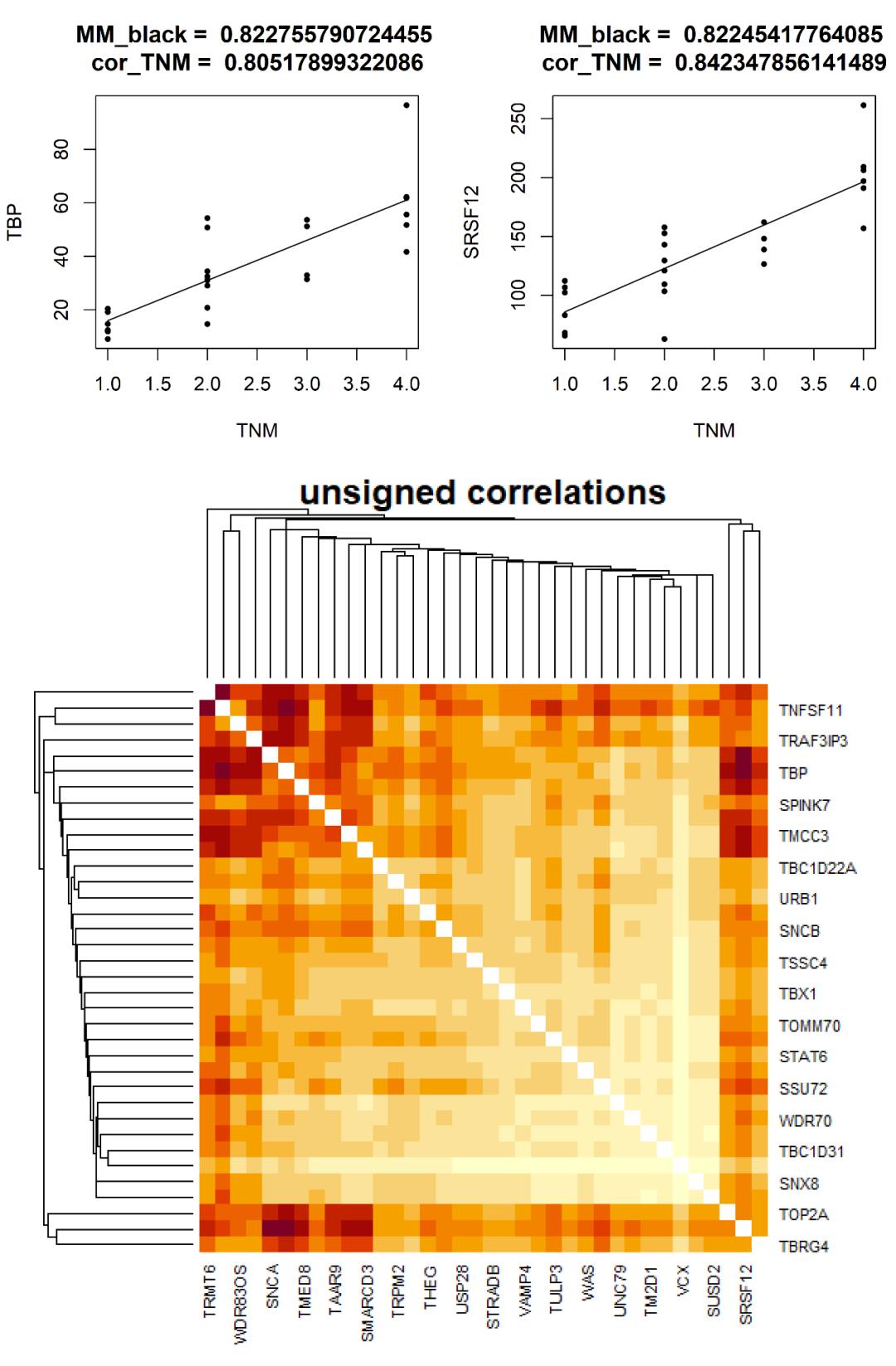
#候选基因与 TNM 分期的相关性散点图
dir.create('black_TNM_cor', recursive = TRUE)
for (i in rownames(select)) {
png(paste('black_TNM_cor/', i, '-TNM.png', sep = ''),
width = 4, height = 4, res = 400, units = 'in', type = 'cairo')
plot(trait[ ,'TNM'], gene[ ,i], xlab = 'TNM', ylab = i,
main = paste('MM_black = ', select[i,'MMblack'], ' cor_TNM = ', select[i,'TNM']), cex = 0.8, pch = 20)
fit <- lm(gene[ ,i]~trait[ ,'TNM'])
lines(trait[ ,'TNM'], fitted(fit))
dev.off()
}
#候选基因间的 TOM 矩阵
plotNetworkHeatmap(gene,
plotGenes = rownames(select),
networkType = 'unsigned',
useTOM = TRUE,
power = powers,
main = 'unsigned correlations')
关于网络可视化
执行上述一系列分析后,也能大致理解了,WGCNA的主要侧重点其实是识别感兴趣的基因集(共表达模块)。
对于网络的可视化是次要内容,但如果有需要,可以将相应的结果导出为的形式,后续再读入至Cytoscape等网络可视化软件中进行共表达网络的可视化。
#输出各模块子网络的边列表和节点列表,后续可导入 cytoscape 绘制网络图
#其中,边的权重为 TOM 矩阵中的值,记录了基因间共表达相似性
dir.create('cytoscape', recursive = TRUE)
for (mod in 1:nrow(table(mergedColors))) {
modules <- names(table(mergedColors))[mod]
probes <- colnames(gene)
inModule <- (mergedColors == modules)
modProbes <- probes[inModule]
modGenes <- modProbes
modtom_sim <- tom_sim[inModule, inModule]
dimnames(modtom_sim) <- list(modProbes, modProbes)
outEdge <- paste('cytoscape/', modules, '.edge_list.txt',sep = '')
outNode <- paste('cytoscape/', modules, '.node_list.txt', sep = '')
exportNetworkToCytoscape(modtom_sim,
edgeFile = outEdge,
nodeFile = outNode,
weighted = TRUE,
threshold = 0.3, #该参数可控制输出的边数量,过滤低权重的边
nodeNames = modProbes,
altNodeNames = modGenes,
nodeAttr = mergedColors[inModule])
}
Cytoscape可视化不在本篇范畴内,还请自行了解了。
参考文献
以上是关于R语言 | 加权基因共表达网络分析(WGCNA)的主要内容,如果未能解决你的问题,请参考以下文章