R语言完美重现STAMP结果图
Posted 宏基因组
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言完美重现STAMP结果图相关的知识,希望对你有一定的参考价值。
写在前面
之前写过一篇关于统计学软件STAMP的教程,使用STAMP对微生物群落数据进行统计学分析还是挺方便的,尤其是对R语言不是很熟悉的朋友来说,图形化的界面和相对简单的操作还是挺友好的。
STAMP——微生物组间差异统计分析
我们通常使用的STAMP的结果主要就是两组数据之间差异性检验的被称作Extended error bar(扩展柱状图)的图像。
由于STAMP的结果图相对固定,可修改的图像参数有限,经常会遇到一些问题,比如靶标物种或功能基因名字过程就会导致显示不全,在与其它图像拼接成一副图的时候也会出现字号太小导致看不清楚的问题。
正好前几天在群里有人询问了这个图有没有其它的绘制办法,今天就给大家带来一个使用ggplot2绘制Extended error bar的方法。
数据准备
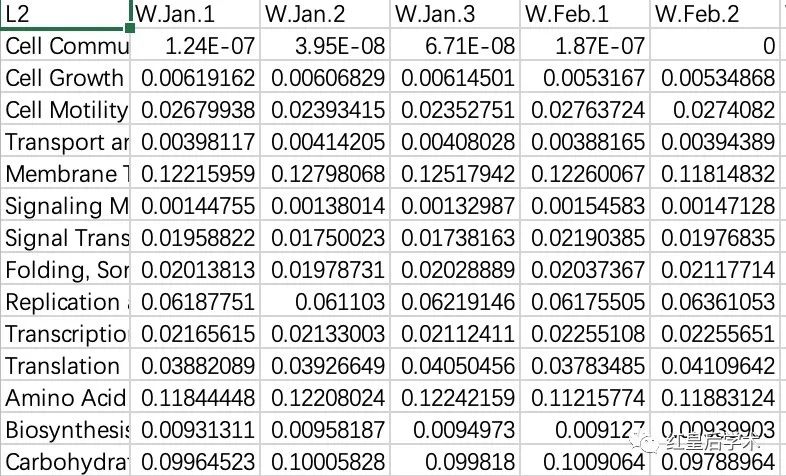
这里我将使用一套同一环境位点水体和沉积物16S扩增子测序的PICRUSt功能预测结果作为示例。
选择的是KEGG L2水平的功能预测的相对丰度数据。
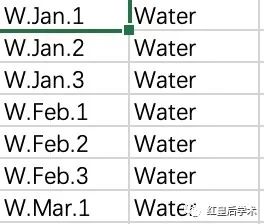
绘图的数据文件有两个,一个是丰度数据,另一个是样本分组数据。
后台回复“STAMP”获取示例文件和完整代码。
首先将数据导入R环境,我是首先过滤掉了平均丰度低于1%的功能分类。
data <- read.table("KEGG_L2.txt",header = TRUE,row.names = 1,sep = " ")
group <- read.table("group.txt",header = FALSE,sep = " ")
library(tidyverse)
data <- data*100
data <- data %>% filter(apply(data,1,mean) > 1)⚠️如果使用不同分类学水平的微生物数据或着更深层次的功能注释数据,由于物种或功能基因种类较多,可能会导致结果中具有差异的数目特别多,比如几十个差异物种放在一副图里基本上是不可能看清的,这种时候就要对数据进行过滤,去除低丰度的物种或基因,具体的过滤标准请根据自身数据情况自行确定。
统计学检验
在绘制Extended error bar之前首先要对数据进行差异显著性检验,以选出丰度在不同组间具有显著差异的物种或功能基因,这里以两组数据为例,使用的检验方式通常为t-test和Wilcox秩和检验。
当分析数据符合正态分布时,使用t-test,如不符合正态分布,则使用Wilcox秩和检验。
t-test
首先对数据进行调整,构建用于t-test的数据框。
data <- t(data)
data1 <- data.frame(data,group$V2)
colnames(data1) <- c(colnames(data),"Group")
data1$Group <- as.factor(data1$Group)由于R语言的t-test一次只能分析一列数据,在网上搜到了一个批量进行t-test的方法,感觉是最简便的了。
首先使用select_if选择格式为数字列,然后使用map_df分别对每一个列进行t-test,最后使用broom:tidy将结果整合在tidy的数据框中。
diff <- data1 %>%
select_if(is.numeric) %>%
map_df(~ broom::tidy(t.test(. ~ Group,data = data1)), .id = 'var')最后对t-test的p值进行校正,保留校正后p值小于0.05的数据。
diff$p.value <- p.adjust(diff$p.value,"bonferroni")
diff <- diff %>% filter(p.value < 0.05)秩和检验
秩和检验和上面的t-test一样,只需要把代码中的t.test换成wilcox.test就可以了。
diff1 <- data1 %>%
select_if(is.numeric) %>%
map_df(~ broom::tidy(wilcox.test(. ~ Group,data = data1)), .id = 'var')
diff1$p.value <- p.adjust(diff1$p.value,"bonferroni")
diff1 <- diff %>% filter(p.value < 0.05)数据可视化
这个Extended error bar的结果图整体分为两个部分,左侧为组建物种或基因丰度平均值的比较条形图,右侧为组间平均丰度及其95%置信区间的散点图。
画图的思路是首先分别绘制左右两幅图,之后再拼接在一起,因此需要首先构建绘制两幅图所需的绘图文件。
绘图数据获取
对于左侧的组间丰度均值比较条形图,我们首先根据差异性检验的结果从原始的丰度数据文件中提取具有显著差异的列,之后按照分组计算其组内平均丰度,再转换成ggplot绘图所用的长格式数据框。
abun.bar <- data1[,c(diff$var,"Group")] %>%
gather(variable,value,-Group) %>%
group_by(variable,Group) %>%
summarise(Mean = mean(value))对于右侧散点图所需要的数据,在上一步差异学检验的结果中已经给出了,我们只需要提取对应的数据列,之后根据差异的正负给其赋予对应的分组信息,最后按照差异丰度的大小对数据进行重新的排序
diff.mean <- diff[,c("var","estimate","conf.low","conf.high")]
diff.mean$Group <- c(ifelse(diff.mean$estimate >0,levels(data1$Group)[1],
levels(data1$Group)[2]))
diff.mean <- diff.mean[order(diff.mean$estimate,decreasing = TRUE),]左侧条形图
在绘制条形图之前,要把物种或基因名按照其组间丰度差异从大到小进行一个排序,以便图像的美观。
在画图之前首先要根据物种或基因丰度的差异大小,对变量的名称设置一个因子并排序,以保证两幅图变量排序的一致性。
library(ggplot2)
cbbPalette <- c("#E69F00", "#56B4E9")
abun.bar$variable <- factor(abun.bar$variable,levels = rev(diff.mean$var))接下来绘制图像。
p1 <- ggplot(abun.bar,aes(variable,Mean,fill = Group)) +
scale_x_discrete(limits = levels(diff.mean$var)) +
coord_flip() +
xlab("") +
ylab("Mean proportion (%)") +
theme(panel.background = element_rect(fill = 'transparent'),
panel.grid = element_blank(),
axis.ticks.length = unit(0.4,"lines"),
axis.ticks = element_line(color='black'),
axis.line = element_line(colour = "black"),
axis.title.x=element_text(colour='black', size=12,face = "bold"),
axis.text=element_text(colour='black',size=10,face = "bold"),
legend.title=element_blank(),
legend.text=element_text(size=12,face = "bold",colour = "black",
margin = margin(r = 20)),
legend.position = c(-1,-0.1),
legend.direction = "horizontal",
legend.key.width = unit(0.8,"cm"),
legend.key.height = unit(0.5,"cm"))for (i in 1:(nrow(diff.mean) - 1))
p1 <- p1 + annotate('rect', xmin = i+0.5, xmax = i+1.5, ymin = -Inf, ymax = Inf,
fill = ifelse(i %% 2 == 0, 'white', 'gray95'))
p1 <- p1 +
geom_bar(stat = "identity",position = "dodge",width = 0.7,colour = "black") +
scale_fill_manual(values=cbbPalette)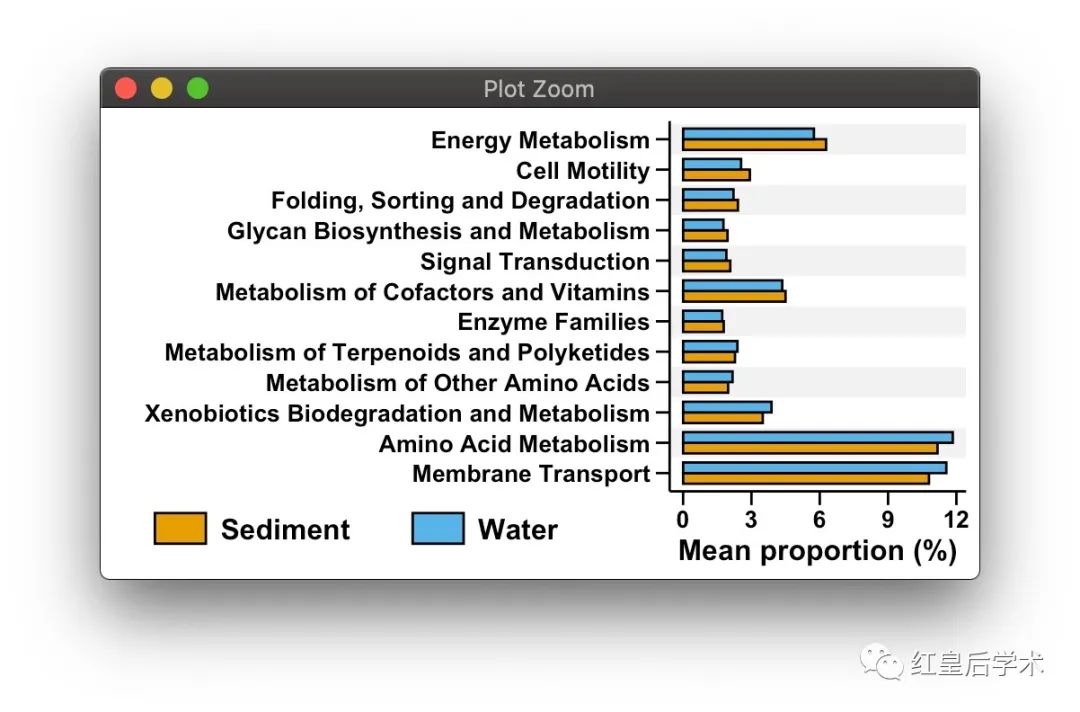
本来是不想用循环添加矩形的方式来实现灰白较差的底纹的,但是研究了半天也没找到更方便的办法,各位要是有更简便的办法欢迎私聊~~
这里要特别注意图层的顺序,一定要在添加举行之后在绘制条形图,不然会覆盖掉。
右侧散点图
同样要先对变量名进行一个因子排序,保证两个图像的变量顺序的一致。
此外散点图要在右侧添加校正后的p-value,因此需要先对p-value取有效数字,之后转换成文本。
diff.mean$var <- factor(diff.mean$var,levels = levels(abun.bar$variable))
diff.mean$p.value <- signif(diff.mean$p.value,3)
diff.mean$p.value <- as.character(diff.mean$p.value)本来是想画散点图的同时直接把p-value的文本加上,但是由于这幅图p-value的文本是添加在绘图区以外的,研究了半天没弄明白,后来给分成了两个图,一副图只有散点图,另一副图只有右侧的p-value文本。
散点图的绘制。
p2 <- ggplot(diff.mean,aes(var,estimate,fill = Group)) +
theme(panel.background = element_rect(fill = 'transparent'),
panel.grid = element_blank(),
axis.ticks.length = unit(0.4,"lines"),
axis.ticks = element_line(color='black'),
axis.line = element_line(colour = "black"),
axis.title.x=element_text(colour='black', size=12,face = "bold"),
axis.text=element_text(colour='black',size=10,face = "bold"),
axis.text.y = element_blank(),
legend.position = "none",
axis.line.y = element_blank(),
axis.ticks.y = element_blank(),
plot.title = element_text(size = 15,face = "bold",colour = "black",hjust = 0.5)) +
scale_x_discrete(limits = levels(diff.mean$var)) +
coord_flip() +
xlab("") +
ylab("Difference in mean proportions (%)") +
labs(title="95% confidence intervals")
for (i in 1:(nrow(diff.mean) - 1))
p2 <- p2 + annotate('rect', xmin = i+0.5, xmax = i+1.5, ymin = -Inf, ymax = Inf,
fill = ifelse(i %% 2 == 0, 'white', 'gray95'))
p2 <- p2 +
geom_errorbar(aes(ymin = conf.low, ymax = conf.high),
position = position_dodge(0.8), width = 0.5, size = 0.5) +
geom_point(shape = 21,size = 3) +
scale_fill_manual(values=cbbPalette) +
geom_hline(aes(yintercept = 0), linetype = 'dashed', color = 'black')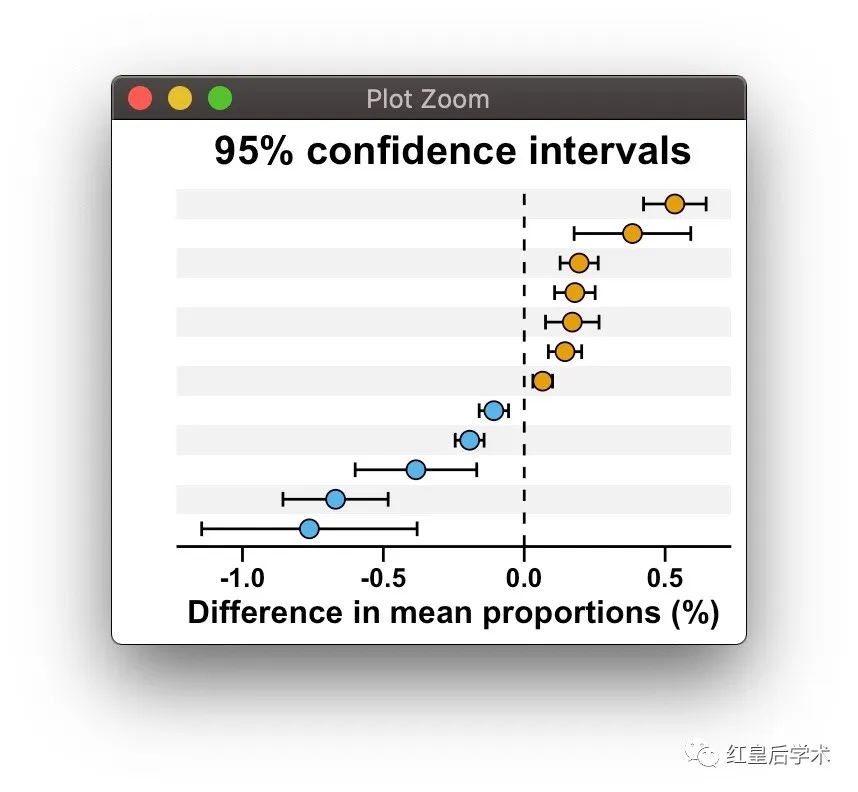
p-value文本的绘制。
p3 <- ggplot(diff.mean,aes(var,estimate,fill = Group)) +
geom_text(aes(y = 0,x = var),label = diff.mean$p.value,
hjust = 0,fontface = "bold",inherit.aes = FALSE,size = 3) +
geom_text(aes(x = nrow(diff.mean)/2 +0.5,y = 0.85),label = "P-value (corrected)",
srt = 90,fontface = "bold",size = 5) +
coord_flip() +
ylim(c(0,1)) +
theme(panel.background = element_blank(),
panel.grid = element_blank(),
axis.line = element_blank(),
axis.ticks = element_blank(),
axis.text = element_blank(),
axis.title = element_blank())
这样所有的绘图单元就齐了。
图像拼接
使用patchwork对3个绘图部分进行拼接。
library(patchwork)
p <- p1 + p2 + p3 + plot_layout(widths = c(4,6,2))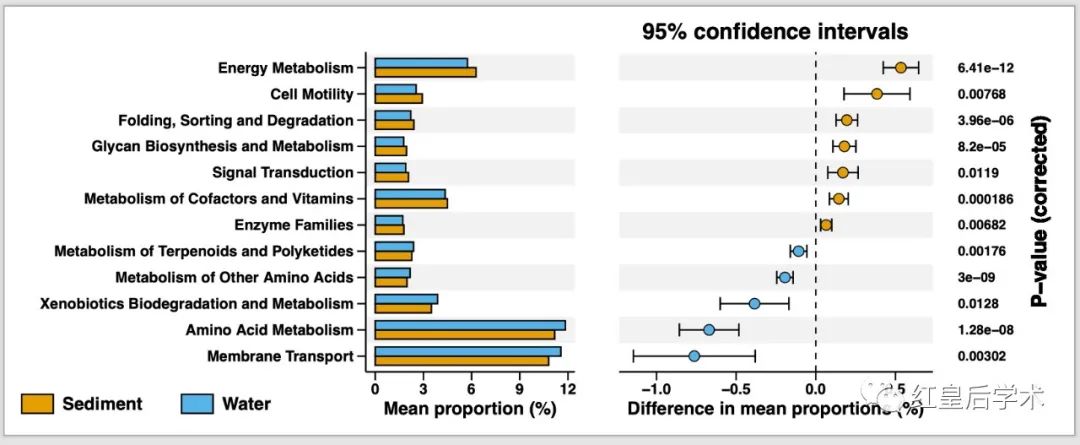
最后保存图片就齐活了。
ggsave("stamp.pdf",p,width = 10,height = 4)后台回复“STAMP”获取示例文件和完整代码。
本文中差异性检验的代码参考了:https://www.it1352.com/1581321.html
另外感谢“生信小白鱼”提供循环添加矩形的代码!!
猜你喜欢
10000+:
系列教程:
专业技能:
一文读懂:
必备技能:
文献阅读
扩增子分析:
在线工具:
科研经验:
编程模板:
生物科普:
写在后面
学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
点击阅读原文,跳转最新文章目录阅读
以上是关于R语言完美重现STAMP结果图的主要内容,如果未能解决你的问题,请参考以下文章