R语言实现分子信息获取
Posted R语言交流中心
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言实现分子信息获取相关的知识,希望对你有一定的参考价值。
我们在前面曾讲到,今天给大家介绍下,读取后如何获取分子的相关信息。Chemistry Development Kit (CDK)数据资源是一个主要获取化学分子属性信息的JAVA库。其官方主页:https://cdk.github.io/。
同时其提供了相对应的JAVA接口供各用户使用。今天就给大家介绍下在R语言中是如何利用其接口进行相应的化合物数据获取的。
首先,我们看下需要安装的包:
install.packages('rcdk')
接下来我们直接通过实例来看下此包的使用:
1. 数据的输入与输出
##数据载入Dir1=system.file("molfiles","kegg.sdf", package = "rcdk")Dir2=system.file("molfiles","dhfr00008.sdf", package = "rcdk")mols <- load.molecules( c(Dir1, Dir2) )

当单个分子sdf文件太大时,我们为了防止内存溢出,那么我们可以遍历读取:
##遍历读取数据iter <- iload.molecules(Dir1,type='sdf')while(iter$hasNext()) {mol<- iter$nextElem()set.property(mol, 'cdk:Title', 'A')print(get.property(mol,"cdk:Title"))}
smile <- 'c1ccccc1CC(=O)C(N)CC1CCCCOC1'mol <- parse.smiles(smile)[[1]]get.smiles(mol)
##多个SMILE结构数据的读取options("java.parameters"=c("-Xmx4000m"))library(rcdk)for (smile in smiles) {m<- parse.smiles(smile)## perform operations on this molecule.jcall("java/lang/System","V","gc")gc()}
##分子的输出,如果参数together=FALSE,则挨个输出分子到sdf文件write.molecules(mols,filename='mymols.sdf')
2. 分子结构的获取
##SMILE转化为2D坐标m <- parse.smiles('CCC')[[1]]m <- generate.2d.coordinates(m)
get.smiles(m,smiles.flavors(c('CxSmiles')))
get.smiles(m,smiles.flavors(c('CxCoordinates')))
3. 分子结构的可视化
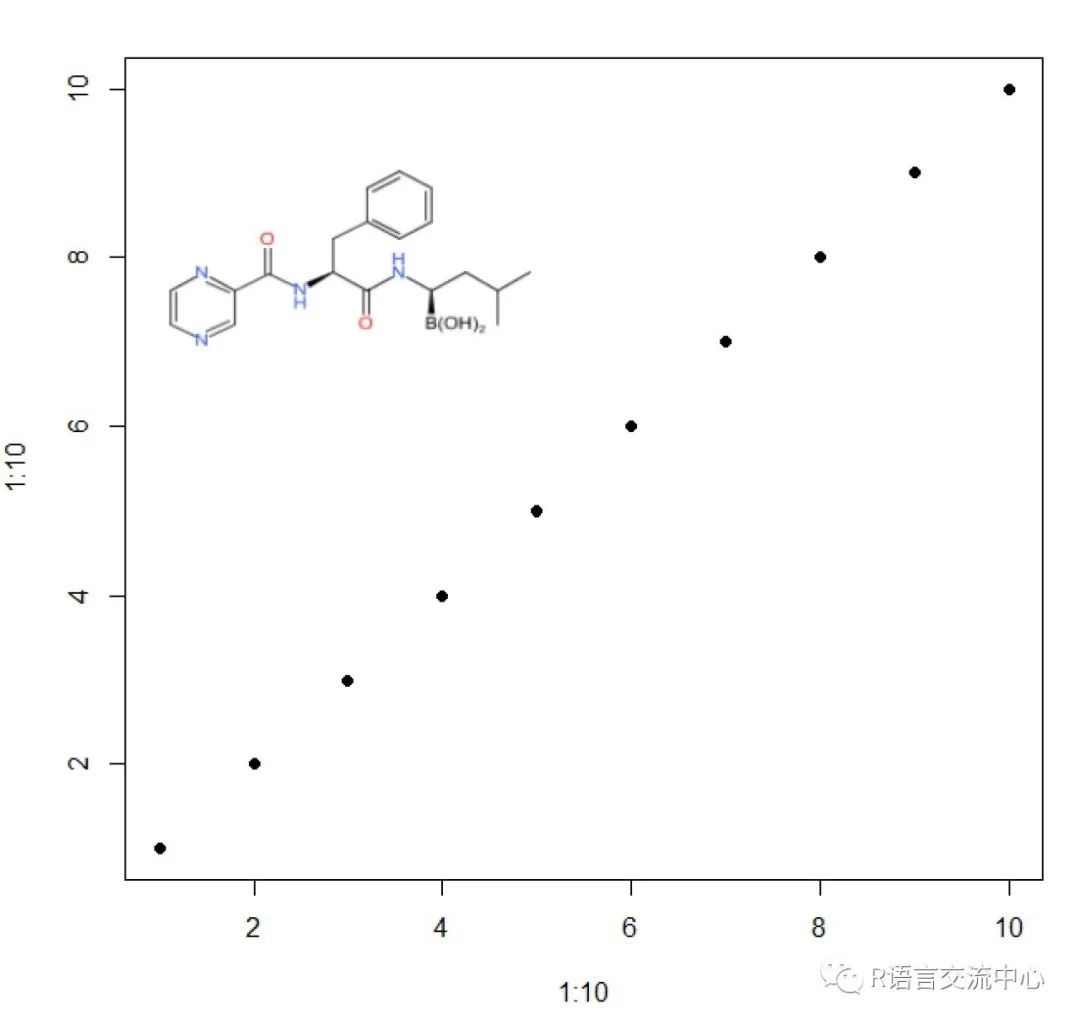
###坐标系中添加分子结构img <-view.image.2d(parse.smiles("B([C@H](CC(C)C)NC(=O)[C@H](CC1=CC=CC=C1)NC(=O)C2=NC=CN=C2)(O)O")[[1]])plot(1:10, 1:10, pch=19)rasterImage(img, 1,6, 5,10)
4. 分子构造的解析
mol <-parse.smiles('c1ccccc1C(Cl)(Br)c1ccccc1')[[1]]atoms <- get.atoms(mol)bonds <- get.bonds(mol)cat('No. of atoms =', length(atoms), ' ')
5. 分子描述信息(此包的核心部分)
#列举此包可获取的分子描述信息属性,包括了拓扑,构造,几何,电子和混合形式。dc <- get.desc.categories()
##获取单个描述的具体信息名称dn <- get.desc.names(dc[4])

###计算描述中的各属性值aDesc <- eval.desc(mol, dn[4])allDescs <- eval.desc(mol, dn)

##获取所有描述属性的集合descNames <-unique(unlist(sapply(get.desc.categories(), get.desc.names)))
###通过描述信息集合获取对应的分子属性data(bpdata)mols <- parse.smiles(bpdata[,1])descNames <- c('org.openscience.cdk.qsar.descriptors.molecular.KierHallSmartsDescriptor','org.openscience.cdk.qsar.descriptors.molecular.APolDescriptor','org.openscience.cdk.qsar.descriptors.molecular.HBondDonorCountDescriptor')descs <- eval.desc(mols, descNames)
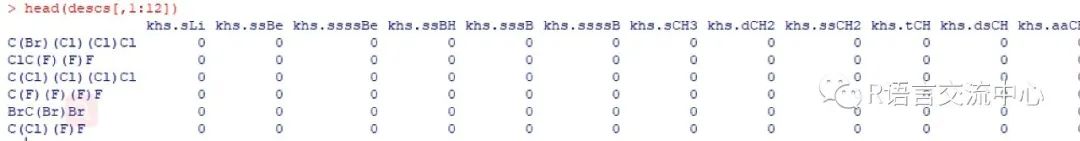
得到上面的矩阵,那么我们就可以进行下面的建模型,分析了。此包也给出了两个例子,我们直接看下:
1. 线性回归模型的构建
###数据清洗descs <- descs[, !apply(descs, 2,function(x) any(is.na(x)) )]descs <- descs[, !apply( descs, 2,function(x) length(unique(x)) == 1 )]r2 <- which(cor(descs)^2 > .6,arr.ind=TRUE)r2 <- r2[ r2[,1] > r2[,2] , ]descs <- descs[, -unique(r2[,2])]
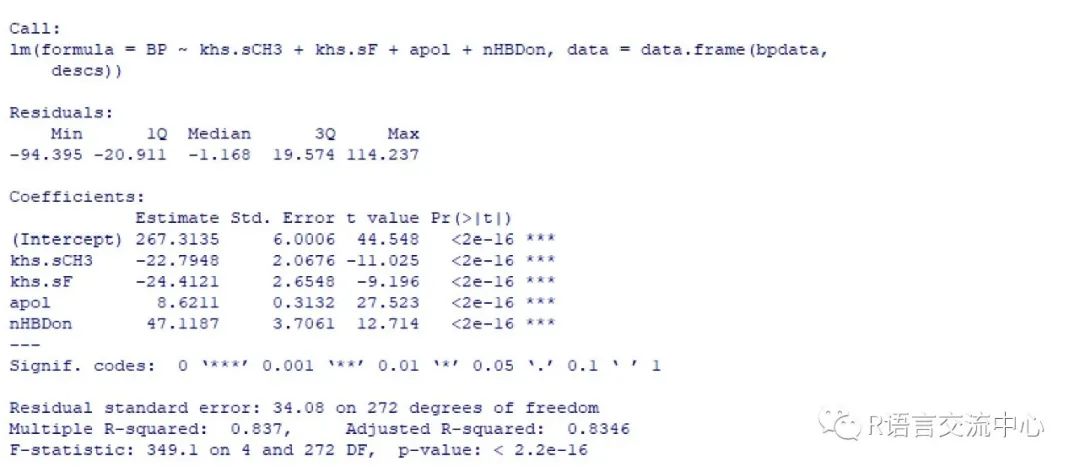
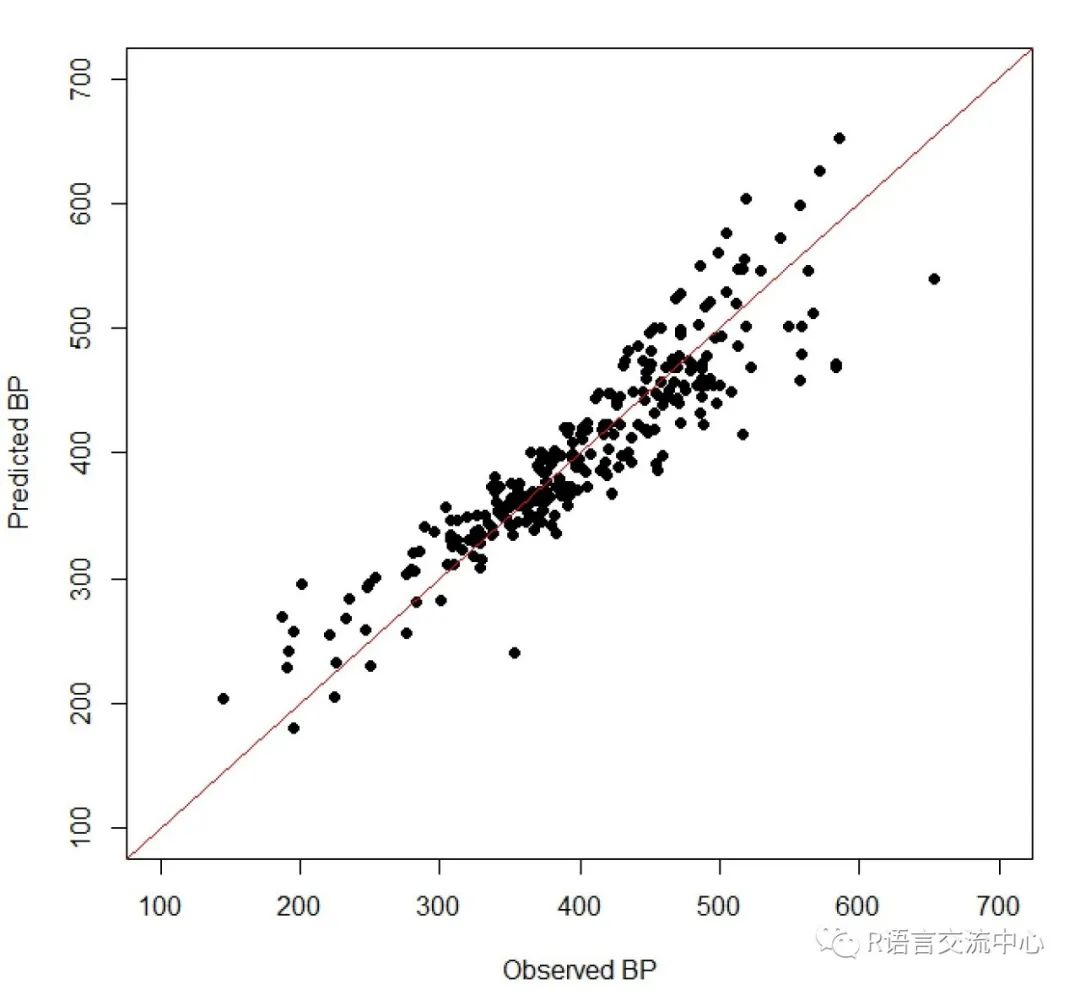
###构建模型model <- lm(BP ~ khs.sCH3 + khs.sF +apol + nHBDon, data.frame(bpdata, descs))summary(model)
##可视化模型预测结果plot(bpdata$BP, predict(model, descs),xlab="Observed BP", ylab="Predicted BP",pch=19, xlim=c(100, 700), ylim=c(100, 700))abline(0,1, col='red')
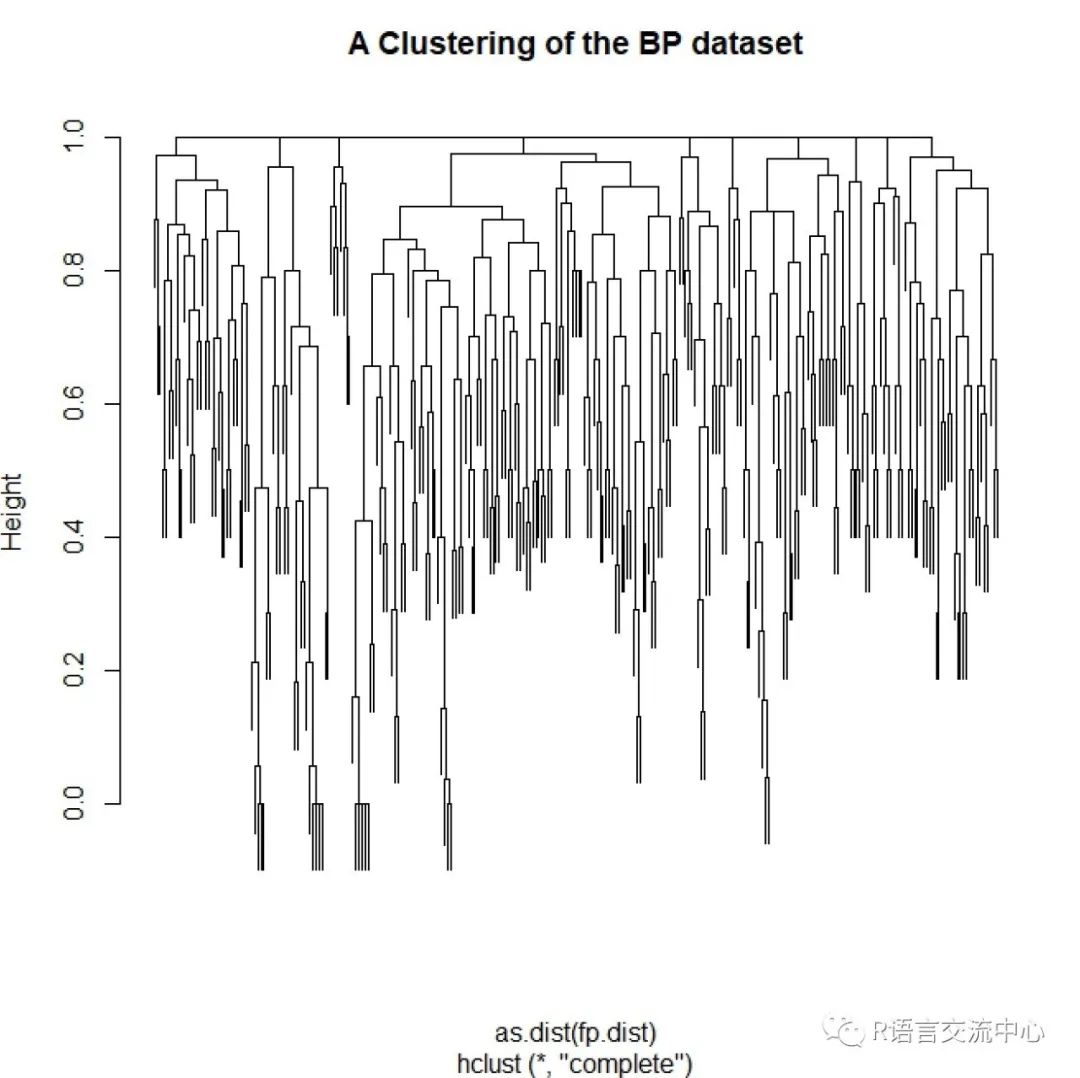
2. 基于分子指纹的分层聚类分析,首先看下分子指纹的类型:

##获取分子指纹,此处类型为circulardata(bpdata)mols <- parse.smiles(bpdata[,1])fps <- lapply(mols, get.fingerprint,type='circular')
##计算距离矩阵fp.sim <-fingerprint::fp.sim.matrix(fps, method='tanimoto')fp.dist <- 1 - fp.sim
##可视化结果cls <- hclust(as.dist(fp.dist))plot(cls, main='A Clustering of the BPdataset', labels=FALSE)
当然,分子指纹还有另外一个功能,那就是分子检索,通过指纹的相似的进行分子的检索,我们直接看下实例:
query.mol <- parse.smiles('CC(=O)')[[1]]target.mols <- parse.smiles(bpdata[,1])query.fp <- get.fingerprint(query.mol,type='circular')target.fps <- lapply(target.mols,get.fingerprint, type='circular')sims <- data.frame(sim=do.call(rbind,lapply(target.fps,fingerprint::distance,fp2=query.fp, method='tanimoto')))subset(sims, sim >= 0.3)

欢迎大家学习交流!
以上是关于R语言实现分子信息获取的主要内容,如果未能解决你的问题,请参考以下文章
R语言sys方法:Sys.getenv函数获取R环境变量Sys.getlocale函数获取当前系统本地信息Sys.setlocale函数设置当前系统本地信息
错误记录Flutter 混合开发获取 BinaryMessenger 报错 ( FlutterActivityAndFragmentDelegate.getFlutterEngine() )(代码片段