R语言实现DNA结构预测
Posted R语言交流中心
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言实现DNA结构预测相关的知识,希望对你有一定的参考价值。
大家对DNA应该都有一定的了解,那么DNA同样不仅仅是具有一级结构的碱基序列,而且还具有二级结构(双螺旋),三级结构(超螺旋)的特征。今天给大家介绍一个来预测DNA结构的R包DNAshapeR,其从基因组测序数据中以超高速、高通量的方式预测DNA形状特征。该软件包以核苷酸序列或基因组间隔作为输入,并生成各种图形表示,以供进一步分析。DNA预测使用滑动五聚体窗口,其中512个不同五聚体中的每一个都有独特的结构特征,从而在每个核苷酸位置(周向)定义了小沟宽(MGW),滚动,螺旋桨扭曲(ProT)和螺旋扭曲(HelT)的向量(周 等人,2013)。MGW和ProT定义碱基对参数,而Roll和HelT代表碱基对步长参数。首先我们看下需要安装的包:
BiocManager::install("DNAshapeR")BiocManager::install("BSgenome.Scerevisiae.UCSC.sacCer3")#CpGmethylation此包需要外网BiocManager::install("BSgenome.Hsapiens.UCSC.hg19")#referencegenome此包需要外网BiocManager::install("AnnotationHub")BiocManager::install("Biostrings")
接下来我们直接通过实例来看下包的操作:
1. 序列的结构预测
library(DNAshapeR)fn <- system.file("extdata","CGRsample.fa", package = "DNAshapeR")pred <- getShape(fn)
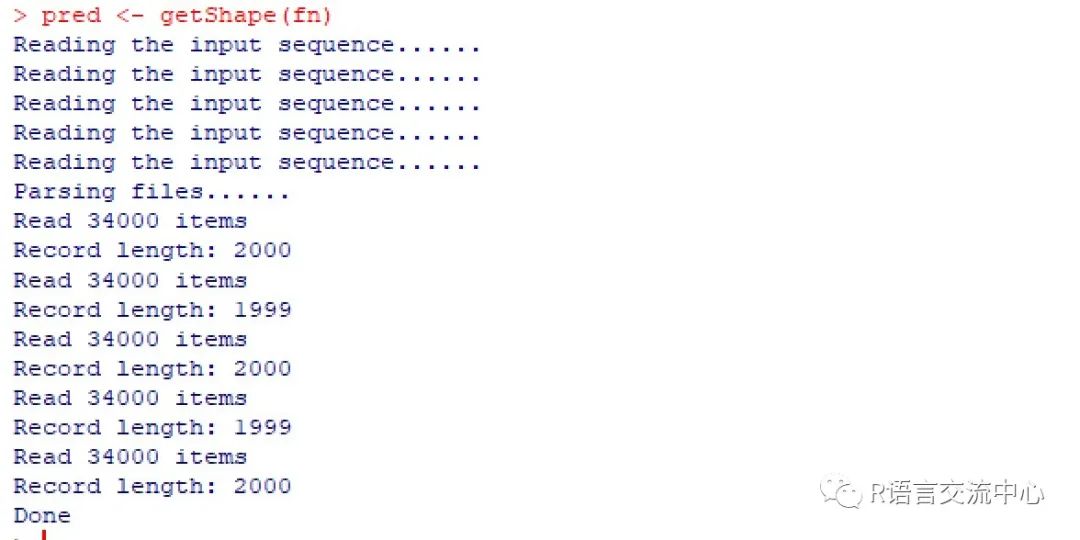
plotShape(pred$MGW)
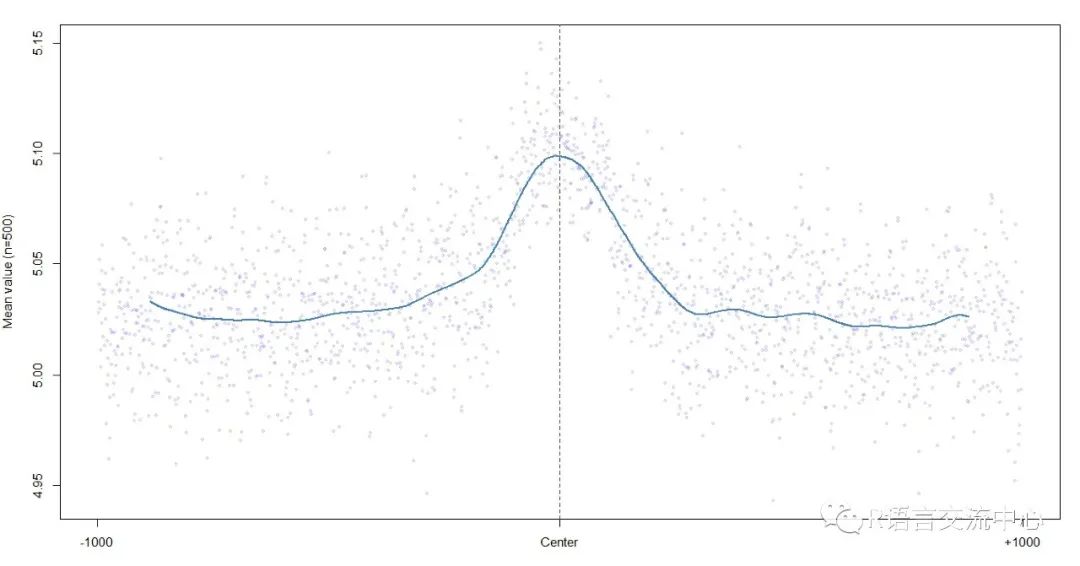
plotShape(pred$ProT)
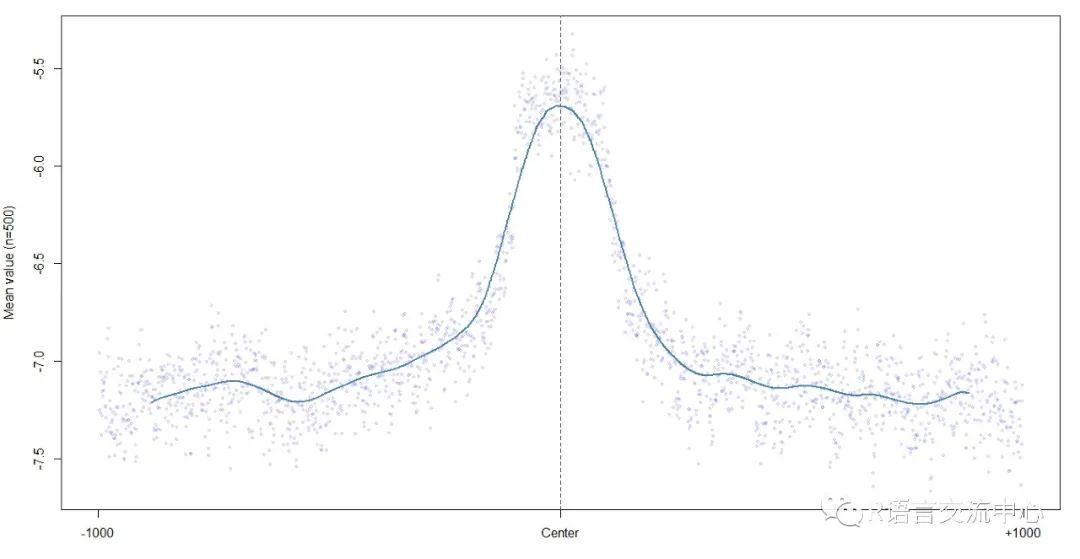
plotShape(pred$Roll)
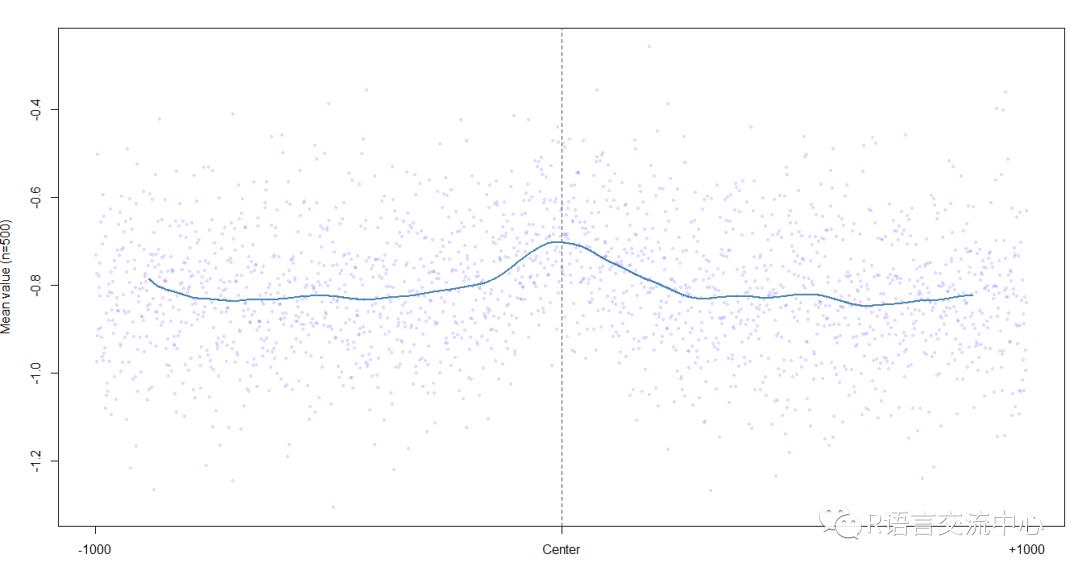
plotShape(pred$HelT) 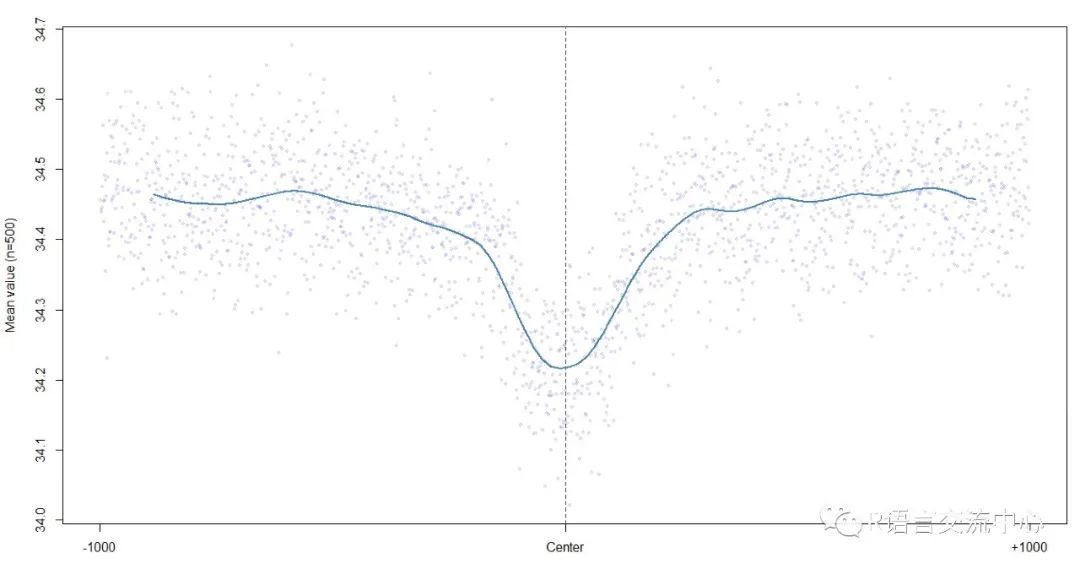
2. 对于参考基因组上的间隔进行预测
library(BSgenome.Scerevisiae.UCSC.sacCer3)# Create a query GRanges objectgr <- GRanges(seqnames =c("chrI"),strand = c("+", "-", "+"),ranges = IRanges(start = c(100, 200, 300), width = 100))getFasta(gr, Scerevisiae, width = 100,filename = "tmp.fa")fn <- "tmp.fa"pred <- getShape(fn)heatShape(pred$ProT, 20)#其中20指的是色块的合并需要的大小即热图的单元格数量
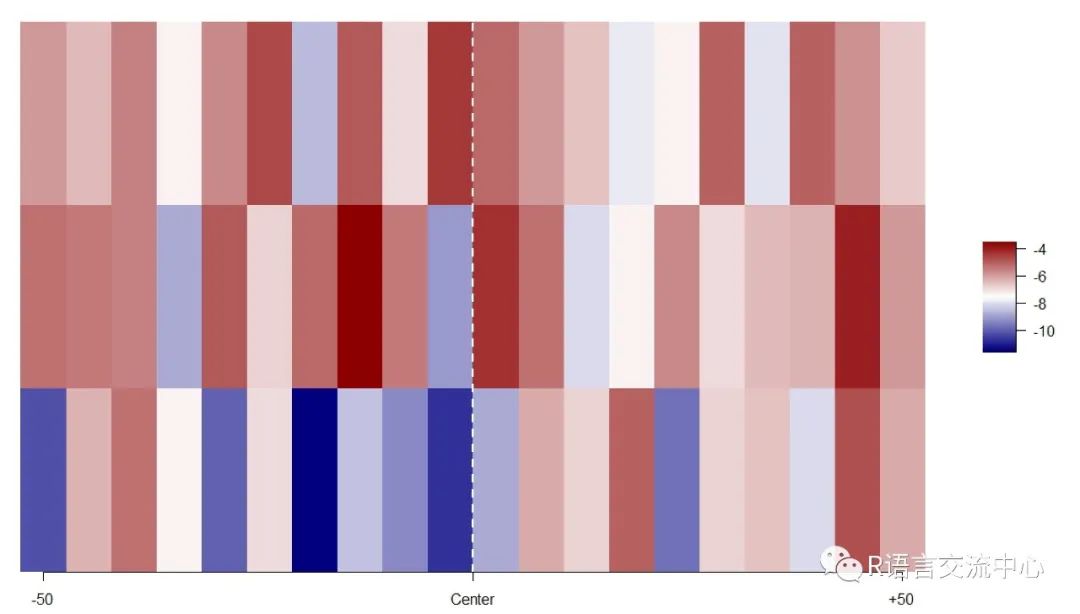
3. 利用公共的平台去检索对应的基因序列做结构预测,当然,我们参考的这个平台的包需要连接外网。
library(BSgenome.Hsapiens.UCSC.hg19)library(AnnotationHub)ah <- AnnotationHub()
ah <- subset(ah, species=="Homosapiens")ah <- query(ah, c("H3K4me3","Gm12878", "Roadmap"))getFasta(ah[[1]], Hsapiens, width = 150,filename = "tmp.fa")

fn <- "tmp.fa"pred <- getShape(fn)
4. 预测CpG甲基化背景下的DNA形态特征,可以制备一个FASTA序列文件,其中符号Mg: M表示前导链上甲基化CpG的胞嘧啶,符号g表示后随链上甲基化CpG的胞嘧啶。例如,
seq1GTGTCACMgCGTCTATACG
library(DNAshapeR)fn_methy <- system.file("extdata","MethylSample.fa", package = "DNAshapeR")pred_methy <- getShape(fn_methy,methylate = TRUE)
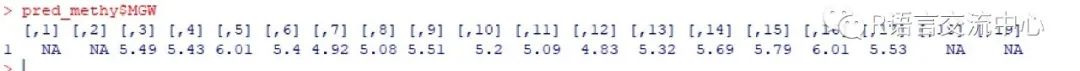
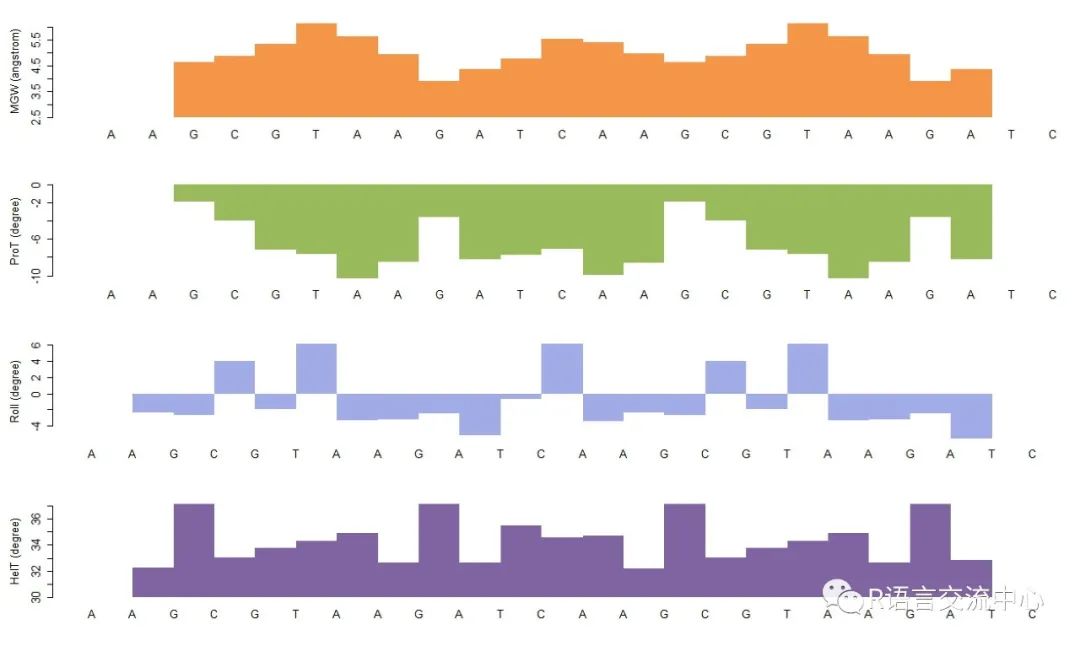
5. 基因组浏览器的类似功能,展示DNA形状的预测结构
fn2 <- system.file("extdata","SingleSeqsample.fa", package = "DNAshapeR")pred2 <- getShape(fn2)trackShape(fn2, pred2) # Only for singlesequence file#如上图,横轴是序列中的每一个碱基的序列,纵轴是对每个点的预测值。
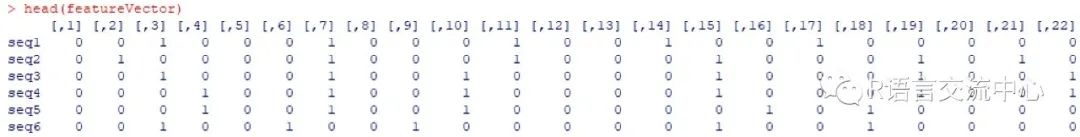
6. DNA序列的编码
library(Biostrings)fn3 <- system.file("extdata","PBMsample_short.fa", package = "DNAshapeR")pred3 <- getShape(fn3)featureType <- c("1-mer","1-shape")featureVector <- encodeSeqShape(fn3,pred3, featureType)head(featureVector)
得到DNA序列的编码,后面具体如何应用那就很广了,在此不再赘述。
欢迎大家学习交流!
以上是关于R语言实现DNA结构预测的主要内容,如果未能解决你的问题,请参考以下文章