利用R语言及perl语言提取基因CDS区实战
Posted 医知圈
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了利用R语言及perl语言提取基因CDS区实战相关的知识,希望对你有一定的参考价值。
医知圈
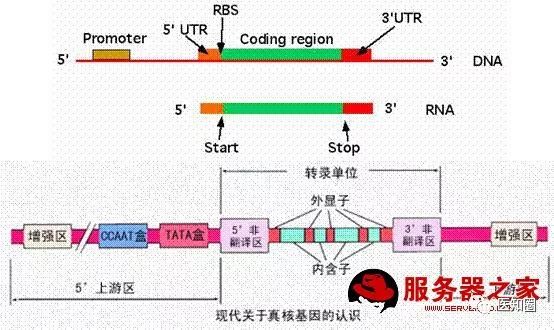
最近挺忙,好久没写了,今天来给大家介绍下利用R语言及perl语言提取基因CDS区。为何要提取CDS区,因为我们在做基因进化分析时,特别是病毒相关的,都要用到,而手工提取太麻烦,今天我给大学分享一下R语言及perl语言代码脚本提取。
R语言代码如下 :
## ---------------------------------------------------------------------
## 1. A TOY EXAMPLE
## ---------------------------------------------------------------------
library(Biostrings)
## A chromosome of length 30:
x <- DNAString("ATTTAGGACACTCCCTGAGGACAAGACCCC")
## 2 transcripts on 'x':
tx1 <- IRanges(1, 8) # 1 exon
tx2 <- c(tx1, IRanges(12, 30)) # 2 exons
transcripts <- IRangesList(tx1=tx1, tx2=tx2)
extractTranscriptSeqs(x, transcripts)
## By default, all the exons are considered to be on the plus strand.
## We can use the 'strand' argument to tell extractTranscriptSeqs()
## to extract them from the minus strand.
## Extract all the exons from the minus strand:
extractTranscriptSeqs(x, transcripts, strand="-")
## Note that, for a transcript located on the minus strand, the exons
## should typically be ordered by descending position on the reference
## genome in order to reflect their rank in the transcript:
extractTranscriptSeqs(x, IRangesList(tx1=tx1, tx2=rev(tx2)), strand="-")
## Extract the exon of the 1st transcript from the minus strand:
extractTranscriptSeqs(x, transcripts, strand=c("-", "+"))
## Extract the 2nd exon of the 2nd transcript from the minus strand:
extractTranscriptSeqs(x, transcripts, strand=list("-", c("+", "-")))
## ---------------------------------------------------------------------
## 2. A REAL EXAMPLE
## ---------------------------------------------------------------------
## Load a genome:
library(BSgenome.Hsapiens.UCSC.hg19)
genome <- BSgenome.Hsapiens.UCSC.hg19
## Load a TxDb object:
txdb_file <- system.file("extdata", "hg19_knownGene_sample.sqlite",
package="GenomicFeatures")
txdb <- loadDb(txdb_file)
## Check that 'txdb' is based on the hg19 assembly:
txdb
## Extract the exon ranges grouped by transcript from 'txdb':
transcripts <- exonsBy(txdb, by="tx", use.names=TRUE)
## Extract the transcript sequences from the genome:
tx_seqs <- extractTranscriptSeqs(genome, transcripts)
tx_seqs
## A sanity check:
stopifnot(identical(width(tx_seqs), unname(sum(width(transcripts)))))
## Note that 'tx_seqs' can also be obtained with:
extractTranscriptSeqs(genome, txdb, use.names=TRUE)
## ---------------------------------------------------------------------
## 3. USING extractTranscriptSeqs() TO EXTRACT CDS SEQUENCES
## ---------------------------------------------------------------------
cds <- cdsBy(txdb, by="tx", use.names=TRUE)
cds_seqs <- extractTranscriptSeqs(genome, cds)
cds_seqs
## A sanity check:
stopifnot(identical(width(cds_seqs), unname(sum(width(cds)))))
## Note that, alternatively, the CDS sequences can be obtained from the
## transcript sequences by removing the 5' and 3' UTRs:
tx_lens <- transcriptLengths(txdb, with.utr5_len=TRUE, with.utr3_len=TRUE)
stopifnot(identical(tx_lens$tx_name, names(tx_seqs))) # sanity
## Keep the rows in 'tx_lens' that correspond to a sequence in 'cds_seqs'
## and put them in the same order as in 'cds_seqs':
m <- match(names(cds_seqs), names(tx_seqs))
tx_lens <- tx_lens[m, ]
utr5_width <- tx_lens$utr5_len
utr3_width <- tx_lens$utr3_len
cds_seqs2 <- narrow(tx_seqs[m],
start=utr5_width+1L, end=-(utr3_width+1L))
stopifnot(identical(as.character(cds_seqs2), as.character(cds_seqs)))
## ---------------------------------------------------------------------
## 4. TRANSLATE THE CDS SEQUENCES
## ---------------------------------------------------------------------
prot_seqs <- translate(cds_seqs, if.fuzzy.codon="solve")
## Note that, by default, translate() uses The Standard Genetic Code to
## translate codons into amino acids. However, depending on the organism,
## a different genetic code might be needed to translate CDS sequences
## located on the mitochodrial chromosome. For example, for vertebrates,
## the following code could be used to correct 'prot_seqs':
SGC1 <- getGeneticCode("SGC1")
chrM_idx <- which(all(seqnames(cds) == "chrM"))
prot_seqs[chrM_idx] <- translate(cds_seqs[chrM_idx], genetic.code=SGC1,
if.fuzzy.codon="solve")
perl语言代码如下 :
#!/usr/bin/perl
use strict;
use warnings;
my @allcdss=(2,8,12,16,18,25,28,35,80,90,102,110,120,125);
my @allcuts;
{
local $/ = 100;
my ($min,$max) = (0,0);
my $left = '';
while (defined (my $line = <DATA>)) {
$line =~ s/ //g;
$min = $max - length($left);
$max = $max + length($line);
$line = $left . $line;
print "$min $max ";
my @allcdss_tmp = grep { $_ > $min && $_ <= $max } @allcdss;
if (@allcdss_tmp) {
my $template = cut2fmt($min,@allcdss_tmp);
print $template," ";
my @fields = unpack($template,$line);
if ($allcdss_tmp[-1] != $max) {
$left = pop @fields;
}
push @allcuts,@fields;
} else {
$left = $line;
next;
}
last if $allcdss_tmp[-1] == $allcdss[-1];
}
}
print "@allcuts ";
sub cut2fmt {
my $minpos = shift;
my(@positions) = @_;
print "@positions ";
my $template = ($positions[0] != $minpos) ? 'x' . ($positions[0] - $minpos - 1) : '';
my $cut_string = 1;
my $lastpos = shift @positions;
foreach my $place (@positions) {
if ($cut_string % 2 == 1) {
my $dist = $place - $lastpos;
$template .= ($dist > 0) ? 'A' . ($dist + 1) : '';
} else {
my $dist = $place - $lastpos;
$template .= ($dist > 0) ? 'x' . ($dist - 1) : '';
}
$lastpos = $place;
$cut_string++;
}
$template .= 'A*';
return $template;
}

以上是关于利用R语言及perl语言提取基因CDS区实战的主要内容,如果未能解决你的问题,请参考以下文章