Perl的学习继续~
Posted 生信星球
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了Perl的学习继续~相关的知识,希望对你有一定的参考价值。
 今天是生信星球陪你的第75天
今天是生信星球陪你的第75天
你想找辆共享单车,发现满街都是别家车,没有一辆你能骑。
你想学点生信,搜了“初学者教程”,满眼尽是高大上,没有一句能看懂。
终于你跨越茫茫宇宙,来到生信星球,发现了初学者的新大陆!
. 子程序
之前用过了chomp、print等,perl还能让用户自定义函数,方便利用已有的代码
使用sub定义【不能以数字开头,最好名字和函数功能相关,不要和内置函数重名】
每个子程序都有一个返回值,而运行子程序要的就是返回值
最简单的使用:
#以打印生信星球信息为例
#!/usr/bin/perl -w
use strict; #代码中有不好的编码风格,那么提示编译失败
&print_planet; # &符号可以调用子程序【定义的和系统不冲突时可以省略&,冲突时必须加上&】
sub print_planet {
print "Hello, bioinfoplanet
";
}
&print_planet;
#这样结果就会输出两次"Hello, bioinfoplanet"
子程序的参数与返回值--输入与输出:
上面的简单使用,没有加上任何参数就会输出字符串这样一个标量;
当然,也可以对子程序输入数组、哈希、文件等参数,然后输出标量、数组或者哈希。
默认将子程序的最后一个表达式的结果作为子程序的返回值,如果想返回中间一个表达式,使用 return 函数
这里以一个加法运算来学习:
#!/usr/bin/perl -w
use strict;
sub sum {
my $num1=shift @_; #定义第一个标量变量为@_中取出的第一的第一个元素
my $num2=shift @_;
my $total=$num1+$num2;
return $total;
}
my $total=&sum (3,5);# &sum()括号中的标量会存储到@_这个数组变量中;括号中有几个参数,@_数组中就有几个元素
print "$total
"
统计fasta信息的子程序:
程序思路:输入有一个,gene id与序列文件对应的哈希列表【格式为id=序列文件】。输出要有三列,一列是gene id,一列是基因数量,一列是基因平均长度。
读取fasta文件时,将$_修改分隔符为>,这样一次就能读进来整段序列,将每段序列存储到哈希中,序列的id为哈希的键,序列为值
#!/usr/bin/perl -w
use strict;
open IN,"<$ARGV[0]"; #输入的第一个文件是基因id与序列对应的哈希文件
while <IN> {
chomp;
my ($id, $file)=split( /=/,$_)[0,1]; #哈希文件的格式是id=序列文件,因此用=分隔二者,存到$_,再将他的第一列赋给变量$id,第二列赋给变量$file
my ($gene_num,$gene_len)=&gene_stat ($file) #定义基因数量、长度的变量,子程序是gene_stat,对上面得到的$file也就是序列文件进行操作
print "$id $gene_num $gene_len
";
}
close IN; #while循环是主体,其中包括了子程序
sub gene_stat { #开始定义子程序
my $file=shift @_; #先定义几个变量file就是刚才得到的数组变量@_中元素,定义基因数量、长度初始值为0
my $gene_num=0;
my $gene_len=0;
open FA,"$file"; #将读入的$file序列文件作为FA句柄
while (<FA>){
chomp;
if (/^>/) { #判断读入的文件开头为>,则作为基因数目
$gene_num+=1;
} else {
my $len=length ($_); #否则作为基因序列,统计长度,一会计算平均长度用
$gene_len+=$len
}
}
close FA;
my $avg_len=$gene_len/$gene_num;
return $gene_num, $avg_len;
}
. Perl常见问题:
Perl的编程过程中,会出现各种各样的问题,需要不断调试。有时候很头疼的就是,即使按照别人的程序敲一遍还是会报错
错误一:忘记加分号
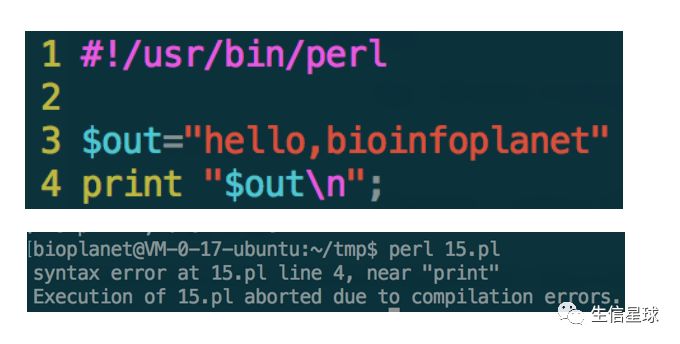
分号在Perl中代表一个完整的语句,忘记加分号就会提示语法错误,并且运行会给出具体出错行数
这里显示第四行有问题,如果在一个大程序中,要快速跳转到某一行,使用vi +4 1.pl ,因为print上一行没有分号
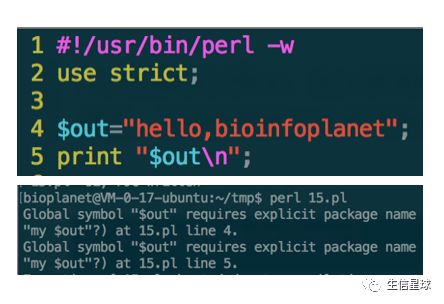
错误二:是否需要用my定义
很多时候需要使用use strict 编译命令,这是就要要使用my 来编译变量,而且不能重复定义
#使用strict的例子:
#!/usr/bin/perl -w
use strict;
my $total;
my $num1=2;
my $num2=1;
$total=$num1+$num2;#这里的total因为之前定义过了,所以不用再使用$total
print "$total
";
错误三:拼写错误
这个在很多编程中都会存在,拼写错误会提示变量没有被声明
这个问题在文本编辑器如Vim中会帮助我们解决,如果某个变量输入错误,比如print输入为prnit ,Vim会通过颜色不同(正确有颜色,错误为白色)
对于变量输入,最好使用vim自带的ctrl+N 或ctrl + P 的自动补全功能,这样可以避免错误
错误四:大括号的使用
尤其使用if嵌套循环时,每一个大括号是一个程序块,如果缺失某一对配对的大括号其中一个,就会提示Missing right/left curly or square bracket at line xxx ;如果大括号多了一个,会提示Unmatched right/left curly bracket at line xxx
避免报错小技能:
perl -c(check)运行之前先检查一下是否书写有问题perl- d(debug)一种交互的perl编程模式,适用于长的脚本。l列出命令的10行;如果要列出某几行,就用l 4-7这样的l+行号
详细信息可以参考perldoc perldebug
. 正则表达式:重要的特性!
Perl的正则表达式是内建的,为Perl提供了快速、灵活的字符串处理
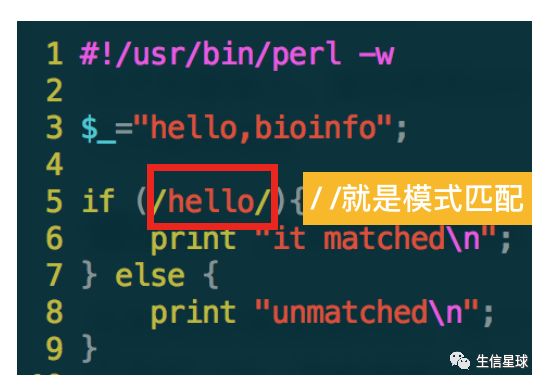
Perl中的正则也叫做“模式匹配”,是用来匹配某字符串的模版;
日常使用中,模式匹配无处不在,例如使用搜索引擎,在搜索框内输入关键字,就能返回信息,他就是利用了正则表达式
一个简单的例子:
元字符:
.替换单个字符(一对一替换),不能替换换行符转义符,例如匹配.或者本身要使用表示数量的
* + ?*【例如在root用户下,使用rm -rf *叫做“自杀模式”】,与.配合使用表示任意字符出现0次或多次 【0—∞】+表示至少一次 【1—∞】?表示0次或1次【范围最小】花括号中指定重复次数,例如匹配1-5次bioinfo中的o
bio{1,5}info如果要制定整个单词次数,用小括号进行分组
/(bioinfo){3}/这个表示匹配bioinfo3次;/(bioinfo)+/表示匹配bioinfo一次以上关于字符集:
匹配字符集的单个字符
/[bcdefwz]at/匹配bat、cat、fat其中一个,等同于/(b|c|d|e|f|w|z)at/或者更简单的/[b-fwz]at/(表示b-f连续的字母加wz)[a-z]、[A-Z]、
[0-9]简写为d+、[a-zA-Z0-9_]也就是单词字符,简写为ws+表示空白,例如split /s+/,$_;反义字符集:利用
^,例如[^0-9]等于[^d]等于D,表示不匹配数字 【反斜线加大些字母表示不匹配】要排除某个匹配,用
!
#来吧!请上手操作一下吧!
#打开你的Linux terminal,新建一个perl脚本
#!/usr/bin/perl -w
use strict;
print "Do you like bioinfoplanet?
"
my $choice=<STDIN>;
if ($choice =~ /y e s/ixs) { #匹配的时候用 =~ 加模式/ /
print "Thanks, good job!
"
} else {
print "OK, we'll do better!
"
}
# 介绍一下$choice =~ /y e s/ixs中的修饰符
# i忽略yes其中各个字母的大小写;
# x可以忽略模式中的空白【常常针对模式较长,都是点号、星号、字符集等,这样写在一起就看着很复杂,需要加空格隔开,但是运行程序的时候又想让他们连在一起,所以用x】
# s可以让点号匹配到换行符,这样即使 "Do you like bioinfoplanet?"这一句是分行显示的,也可以匹配成功
# m:多行匹配,跨行模式匹配
# g全局匹配
生信模式匹配小练习:
想寻找开头是CCG,结尾是TTG的序列
#!/usr/bin/perl -w
use strict;
open IN,"<$ARGV[]0";
while (<IN>) {
chomp;
if (/^CGG.*TTG$/i) { #^表示开头匹配,$表示结尾匹配
print "$_
";
} else {
next;
}
}
close IN;
#补充一个vim小技巧,利用d(删除)再结合^或$,可以表示删除光标到行首/尾
单词锚定:
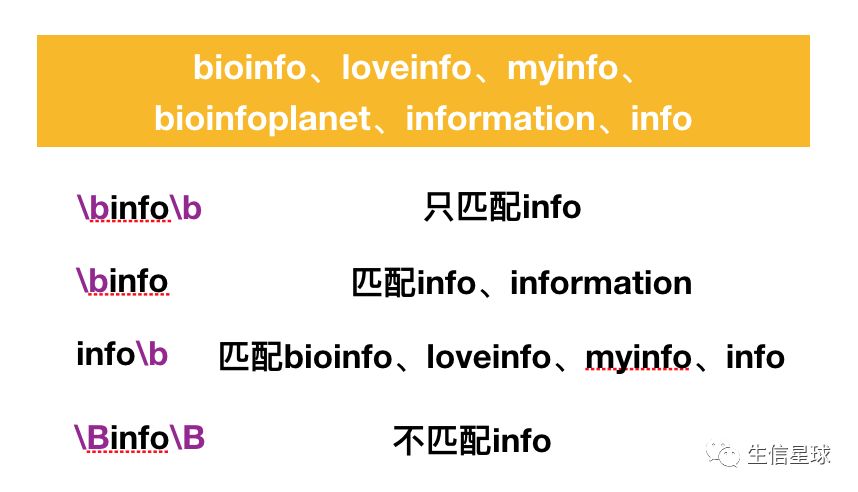
挖掘文本信息:
一般本地blast的format 0结果中,会有匹配的分值(Score)、期望值(Expect)、一致性(Identities)、准确度(Positives)、Gaps等信息,可以挖掘这些信息
#比如这里有一个blast format0的结果,result1中想要数字部分,result2想要括号中的百分数部分
#!/usr/bin/perl -w
use strict;
my $result1="Score = 150 bits (308), Expect = 1e-34, Method: Compositional matrix adjust.";
if ($result1=~ m/Scores*=s*(d+)s*(?:bits|Bits)s*(d+),s*Expects*=s*(S+),.*/) { #匹配过程看起来很复杂,其实也就是一点点填充的过程,有空格就用s*,有数字就用d+,对于中间没有空格的整体如1e-34就直接使用S+。就像这样把整个句子用元字符的方式重绘出来,方便大量统计
print "$1 $2
"; #$1与$2就是捕获之前分组的变量
}
#得到的结果就是 150 1e-34
#按说每一个括号就是一个分组,而后面用$来捕获变量也是根据括号,那么比如(bits|Bits),我们只想让他用来分组,表示匹配bit或者Bits。但是这样有一个问题,后来引用的时候就会讲这一部分也输出;为了避免这个问题,就在左边括号后面加上?:表示这个整体就是一个分组,别把它当变量使用
my $result2="Identities = 131/468 (28%), Posotives = 217/468 (46%), Gaps = 46/468 (10%)";
if ($result2=~ m/(S+)/g) {
print "$result2
";
}
改进版:
#!/usr/bin/perl -w
use strict;
my $result1="Score = 150 bits (308), Expect = 1e-34, Method: Compositional matrix adjust.";
my $result2="Identities = 131/468 (28%), Posotives = 217/468 (46%), Gaps = 46/468 (10%)";
my @info1= ($result1=~ m/Scores*=s*(d+)s*(?:bits|Bits)s*(d+),s*Expects*=s*(S+),.*/);
print "@info1
";
#直接将捕获的变量存储到列表中,即使没有匹配到,也只是会输出一个空列表
my %info2= ($result2=~ m/(S+)/g); #加上g意思是将全部符合条件的找出来
print "@info2
";
另外还有一个s(替换模式),例如
#!/usr/bin/perl -w
use strict;
my $line="hello, bioinfoplanet";
$line=~ s/hello/nihao/;#结果就将hello变成了nihao
$line=~ s/^/Hi,/; #结果为Hi,nihao, bioinfoplanet
$line=~ s/$/hi/; #结果在结尾加上一个hi
$line=~ s/hello//g; #将全部hello替换为空,大规模替换要加上g
print "$line
";
对序列进行处理:
得到序列的反向/互补序列
#!/usr/bin/perl -w
use strict;
my $seq="TTCGATGCTAGCATCGATCGCTAGCATCGCTAGCATCCCGATTGACTAATCAT";
my $qes=reverse $seq;
print "$qes
";#序列反转
$qes=~ tr/ATCGN/TAGCN/; # 序列碱基互补
print "$qes
";
#结果1得到:TACTAATCAGTTAGCCCTACGATCGCTACGATCGCTAGCTACGATCGTAGCTT
#结果2得到:ATGATTAGTCAATCGGGATGCTAGCGATGCTAGCGATCGATGCTAGCATCGAA
序列全变大些/小写
#!/usr/bin/perl -w
use strict;
my $seq="TTCGATGCTAGCATCGATCGCTAGCATCGCTAGCATCCCGATTGACTAATCAT";
$seq =~ s/(w+)/L$1/g; #(w+)是将所有字母分成一组,L是小写[U是大写],$1是捕获前面的分组【另外u是更改首个字母大写;l首字母变小写】
print "$seq
";
#结果得到:ttcgatgctagcatcgatcgctagcatcgctagcatcccgattgactaatcat
另外这些大小写语句除了在s模式下使用,还能在双引号内使用,例如在print 语句中格式化输出print "Uhello, worEld" 输出结果是HELLO, WORld 【注意这里的E是终止前面U命令的意思】
贪婪匹配 -- 匹配尽可能多的内容
一般. + ? 会匹配尽量多的内容,比如上个例子$seq 中,如果匹配模式是$seq=~ /^TTC(.+)CAT/i ,即使中间也出现了CAT,程序也不会停下,他会一直匹配到结尾的CAT,这就是贪婪;
贪婪的对立面就是节俭,这种模式可以在. + ? 的基础上加上? ,也就是$seq=~ /^TTC(.+?)CAT/i 就输出最短的序列
格式化序列
有时一个fasta文件中序列长短不一,并且有的换行有的不换行,这样看起来就很难看。这时就可以将序列格式化,让每行保证一定量的字符【可以自定义】,并且加换行符。
测试fasta下载:wget https://github.com/Bioinfoplanet/Perl-Test/blob/master/unformated.fasta
#!/usr/bin/perl -w
use strict;
open FA,"<$ARGV[0]";
$/=">";
<FA>;
while (<FA>) {
chomp;
my ($id,$seq)=(split /
/,$_,2)[0,1];
$seq=~ s/
//g;
$seq=~ s/(w{$ARGV[1]})/$1
/g;
print ">$id
$seq"
}
初学生信,很荣幸带你迈出第一步。
我们是生信星球,一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到Bioplanet520@outlook.com~
以上是关于Perl的学习继续~的主要内容,如果未能解决你的问题,请参考以下文章