原核生物蛋白质翻译的起始过程?
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了原核生物蛋白质翻译的起始过程?相关的知识,希望对你有一定的参考价值。
原核生物的蛋白质生物合成氨基酸在核糖体上缩合成多肽链是通过核糖体循环而实现的。此循环可分为肽链合成的起始(intiation),肽链的延伸(elongation)和肽链合成的终止(termination)三个主要过程。原核细胞的蛋白质合成过程以e.coli细胞为例。
肽链合成的起始
1.三元复合物(trimer
complex)的形成核糖体30s小亚基附着于mrna的起始信号部位,该结合反应是由起始因子3(if3)介导的,另外有mg2+的参与。故形成if3-30s亚基-mrna三元复合物。
2.30s前起始复合物(30s
pre-initiation
complex)的形成在起始因子2(if2)的作用下,甲酰蛋氨酸-起始型trna(fmet-trna
met)与mrna分子中的起始密码子(aug或gug)相结合,即密码子与反密码子相互反应。同时if3从三元复合物脱落,形成30s前起始复合物,即if2-30s亚基-mrna-fmet-trnamef复合物。此步亦需要fgtp和mg2+参与。
3.70s起始复合物(70s
initiation
complex)形成。50s亚基与上述的30s前起始复合物结合,同时if2脱落,形成70s起始复合物,即30s亚基-mrna-50s亚基-fmer-trna
met复合物。此时fmet-trna
met占据着50s亚基的肽酰位(peptidyl
site,简称为p位或给位),而50s的氨基酰位(aminoacyl
site,简称为a位或受位)暂为空位。
原核细胞蛋白质合成的起始过程氨基酸活化(fmet-trnamet形成)
关于原核细胞蛋白质生物合成的起始过程讨论以下几点:
1.核糖体亚基(30s和50s):70s核糖体颗粒必须解离为亚基,才能参与形成30s前起始复合物及70s起始复合物,if3除具有形成三元复合物活性外,也具有使70s核糖体颗粒解离为30s和50s亚基的作用,即解离因子活性(disassociation
factor
activity)。
2.if:用高浓度的盐(如0.5mol/lkcl)洗涤核糖体,可使核糖体解离为亚基,但这些核糖体亚基在蛋白质生物合成的起始阶段没有活性。后来从盐洗涤液中分离出三种蛋白质因子,当把这三种因子加入盐洗过的核糖体后,核糖体再现了活性,故将这三种蛋白质因子依次命名为if1、if2和if3.在70s起始复合物形成后,无任何if与之结合。if的生物学活性见下表。值得一提的是if1无特异功能,仅具有加强if2和if3的活性作用,这种广泛的效应亦称为基因多效性。
参考资料:www.37c.com.cn/..._3.htm 参考技术A 氨基酸在核糖体上缩合成多肽链是通过核糖体循环而实现的。此循环可分为肽链合成的起始(intiation),肽链的延伸(elongation)和肽链合成的终止(termination)三个主要过程。原核细胞的蛋白质合成过程以E.coli细胞为例。
肽链合成的起始
1.三元复合物(trimer complex)的形成核糖体30S小亚基附着于mRNA的起始信号部位,该结合反应是由起始因子3(IF3)介导的,另外有Mg2+的参与。故形成IF3-30S亚基-mRNA三元复合物。0994-1a.jpg (10353 bytes)
2.30S前起始复合物(30S pre-initiation complex)的形成在起始因子2(IF2)的作用下,甲酰蛋氨酸-起始型tRNA(fMet-tRNA Met)与mRNA分子中的起始密码子(AUG或GUG)相结合,即密码子与反密码子相互反应。同时IF3从三元复合物脱落,形成30S前起始复合物,即IF2-30S亚基-mRNA-fMet-tRNAMef复合物。此步亦需要fGTP和Mg2+参与。0994-1b.jpg (13753 bytes)
3.70S起始复合物(70S initiation complex)形成。50S亚基与上述的30S前起始复合物结合,同时IF2脱落,形成70S起始复合物,即30S亚基-mRNA-50S亚基-fMer-tRNA Met复合物。此时fMet-tRNA Met占据着50S亚基的肽酰位(peptidyl site,简称为P位或给位),而50S的氨基酰位(aminoacyl site,简称为A位或受位)暂为空位。
原核细胞蛋白质合成的起始过程氨基酸活化(fMet-tRNAMet形成) 参考技术B 1. 核糖体大、小亚基的分离起始因子3(initiation factors,IF-3)3和IF-1和核糖体结合,使核糖体大、小亚基分开,以利于mRNA和fMet-tRNA结合到核糖体小亚基上。
2. mRNA与小亚基结合 原核生物中每一个mRNA的5'-端都具有核糖体结合位点,它是位于AUG上游8~13个核苷酸处由4~6个核苷酸组成的富含嘌呤的序列,又称为SD序列。这段序列正好与30S小亚基中的16s rRNA3’端一部分序列互补,因此SD序列又称为核糖体结合位点(ribosomal binding site,RBS)。紧接SD序列的小段核苷酸又可以被核糖体小亚基蛋白辨认。原核生物就是靠这种核酸-核酸、核酸-蛋白质之间的辨认结合把mRNA结合到核糖体的小亚基上。该结合反应需IF-3、 IF-1的参与。
3. 甲酰甲硫氨酰-tRNA的结合 在IF-2作用下,fMet-tRNA与mRNA分子中的AUG相结合,即密码子与反密码子配对,此步需要GTP和Mg2+参与。
4. 核糖体大小亚基结合 fMet -tRNA结合后,IF-3脱离小亚基,随着IF-3的脱落,核糖体50S大亚基与30S小亚基结合形成70S的起始复合物。与此同时GTP水解,IF-1和IF-2脱离起始复合物,甲酰甲硫氨酰-tRNA占据P位,A位是空的,因此与mRNA上第二个密码子对应的氨基酰-tRNA即可进入A位。本回答被提问者采纳
专家点评Nat Methods | 邢毅团队利用深度学习强化RNA可变剪接分析的准确性
点评丨张强锋(清华大学生命科学学院教授)、马坚(美国卡耐基梅隆大学计算机学院教授)
责编 | Spotlight
真核生物的基因区别于原核生物的一个重要特征是具有内含子,内含子的存在,使得真核基因的表达必须经过RNA剪接这一重要步骤。高等真核生物的一个基因中含有多个内含子。通过对RNA剪接的调控,使得一个基因能够转录出多个不同的转录本,从而编码多个蛋白质,增加了生命体的复杂度和适应度【1】。
剪接反应由剪接体催化完成,剪接体含有170多个相关蛋白,具有高度的复杂性与动态性【2】。虽然通过冷冻电镜技术已经获得了多个酵母及人的剪接体反应中间状态的结构【3】,但是以目前对剪接反应机制的了解,仍然不能根据序列等条件不经过实验而直接较为准确的推断RNA可变剪接的结果。对RNA剪接的研究,还是需要通过实验手段进行,最常用的便是RNA-seq技术。
RNA-seq技术虽然能较好的定量基因表达的结果,但是对于差异剪接分析来说,它需要更高的测序深度。即便如此,现有的计算方法仍然不能较为准确的定量低表达基因的剪接变化。因此,为了提高剪接定量的准确性,急需引入新的计算分析方法。
2019年3月25日,来自宾夕法尼亚大学/费城儿童医院的邢毅教授团队在Nature Methods上发表了题为Deep-learning augmented RNA-seq analysis of transcript splicing的工作,首次将深度学习与贝叶斯假设检验结合,用于RNA可变剪接分析。该文章提出了一种新的计算框架(DARTS,Deep-learning Augmented RNA-seq analysis of Transcript Splicing),通过对公共数据库中大量RNA-seq结果的深度学习,即使对于那些测序深度不那么高的样品,也能够有效提高RNA-seq定量差异剪接的准确度。
DARTS由两部分构成:深度神经网络模块(DNN)和贝叶斯推断模块(BHT)。DNN基于顺式序列特征和样品特异的RNA结合蛋白表达水平特征来预测差异剪接的结果;BHT通过整合实验样品测序数据本身和基于深度神经网络的先验概率来推断差异剪接的结果(图1)。
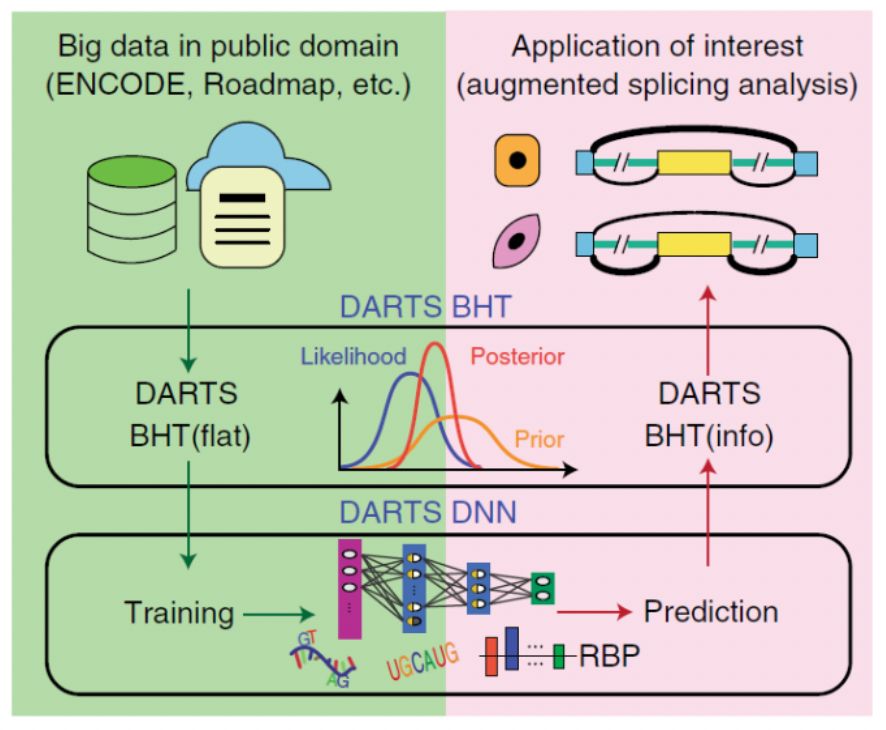
图1. DARTS的基本框架
与别的计算方法不同,DARTS DNN不仅通过顺式序列特征来预测可变剪接的结果,而且还将样品中RNA结合蛋白的表达水平整合进了RNA可变剪接结果的预测中,增加了预测参数的维度(图2)。
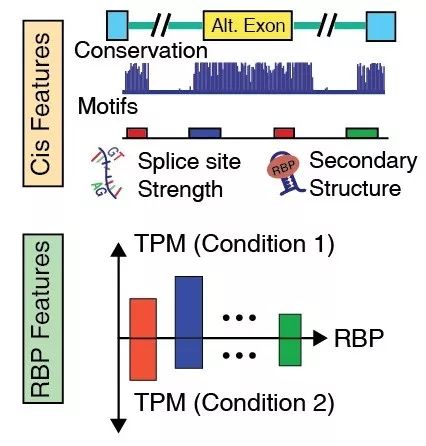 图2. DARTS DNN的特征
图2. DARTS DNN的特征
通过DNN对ENCODE 和Roadmap数据库中大量RNA-seq结果的深度学习,能够获得高精度的预测值作为BHT中的贝叶斯先验概率,进而结合具体实验中RNA-seq的结果,获得更为准确的差异剪接推断。研究者发现,在低通量RNA-seq文库中,通过使用DNN预测值进行强化分析后,能够达到比使用传统方法分析更高的准确度,并且这种提升在越低通量的文库中越发明显。即使在高通量的RNA-seq文库中,使用DNN预测仍能发现在低表达基因中的可变剪接变化。而在过去,这些低表达基因的可变剪接变化在传统分析方法中会被忽略。DARTS不仅提升了基于RNA-seq方法研究可变剪接的准确性,同时也提供了在低表达基因中研究可变剪接的研究手段。
DARTS目前在GitHub上开源了深度学习的预测、训练模块、特征值,以及贝叶斯统计模型(https://github.com/Xinglab/DARTS)。用户可以通过Anaconda实现一键安装DARTS。学术用户可以免费使用DARTS, 商业用户需联系作者获取软件使用授权。
专家解读

张强锋(清华大学生命科学学院教授)
可变剪接现象从70年代发现后,其基本的科学问题聚焦为可变剪接位点发现、差异分析、调控元件和网络的发现和构建。RNA-seq 技术的发明,使得系统、定量的可变剪接差异分析成为可能。大量测序数据的可变剪接差异分析需要优秀的统计模型和计算工具,因此一直是一个需要高度技巧的生物信息学研究课题。宾夕法尼亚大学/费城儿童医院的邢毅教授研究组在此领域深耕多年,贡献了多个有影响力的算法和计算工具。邢毅教授最先提出了用“剪接图”(splicing graph)的数据结构和概率算法来推导和定量转录组的mRNA异构体【4,5】,这些算法在转录组分析领域产生了深刻的影响,已经成为高通量RNA测序数据分析的算法基础。针对高通量RNA-seq数据,邢毅教授研究组开发出的用于差异剪接分析的rMATS软件【6】,已经在全世界被下载超过25,000次,被广泛用于转录组的数据分析。再加上别的研究组开发的例如MISO、SpliceTrap、MAJIQ、SUPPA等软件,虽然各有特点,但对于测序较深、质量较好的数据集都能取得不错的结果。然而,有大量RNA-seq 测序实验在设计时,由于成本等原因,测序深度较浅。对于这些数据集,能利用来做差异分析的可变剪接事件非常有限。
在方法上,DARTS最大的特色是巧妙利用了公开的RNA-seq大数据样本,使用深度神经网络学习得到了外显子差异剪接的贝叶斯假设检验统计模型的准确先验概率分布。另外独立来看,使用深度神经网络通过顺式序列和反式因子RBP表达丰度进行差异剪接预测的思路也值得借鉴。总的来说,DARTS综合了深度学习和贝叶斯假设检验统计模型的优点,为那些低测序深度的数据提供了更好的做可变剪接的分析的手段,拓展了传统RNA-seq可变剪接分析的敏感度和准确度。
马坚(美国卡耐基梅隆大学计算机学院教授)
近些年来深度学习的应用席卷各行各业。在系统生物学领域,尤其是基因组学,由于其海量数据的积累,加上众多奇妙而未知的生物学问题,研究人员对深度学习的使用同样变得如火如荼。但是如何可靠有效的利用深度学习去体现基因组海量数据的潜在优势,并且真正发现新的生物学知识是基因组学和计算生物学最有意义的挑战之一。
邢毅教授实验室的这个DARTS工作清新优雅不落俗套,在目前众多深度学习在基因组学的应用中明显脱颖而出。从计算方法设计的策略和概念角度而言,此工作的最大的亮点是充分利用海量公有数据如ENCODE,但模型本身又不完全依赖于这些公有数据。也就是说,有效综合了海量数据的训练以及新样本的特殊性。DARTS的整体思想是用深度神经网络从现有海量数据中找出通用的有用信息作为先验,然后用贝叶斯假设检验结合来自样本本身的RNA-seq数据信息做可变剪接的预测。在基因组学中,确实有很多类似的问题 -- 如何在现有数据(已有细胞系)上对特定的基因组标注(譬如染色质结构、转录因子结合)训练一个机器学习模型并在完全新的细胞系中有效预测,已经成为一个计算基因组学中广泛关注的问题。因此,DARTS崭新的整体设计理念值得很多其他类似的问题借鉴。
从模型本身的技术角度而言,DARTS有效利用了深度神经网络对异质数据特征的整合,并且整个计算方法的评测和方法比较都明智而审慎。譬如,DARTS的深度神经网络部分结合了剪接位置附近的序列信息、进化信息、可变剪接产生的RNA二级结构信息、RBP的表达信息等。DARTS巧妙的利用深度神经网络预测的结果来作为贝叶斯假设检验中先验,结合样本本身的RNA-seq序列信息实现了更可靠的可变剪接预测。文中其他对于机器学习方法的评测同样可圈可点。例如,常见的对正负样本不均衡的问题对模型训练和评测带来可能的偏差有细致入微的控制。另外,对模型中每个模块的贡献也做了详细分析。随着RNA-seq数据的不断积累,相信DARTS会有广泛的应用,尤其是在RNA-seq测序深度并不高的实验情况下。这个计算工具对进一步理解可变剪接在不同细胞状态下的调控机理将有深远的意义。
常有人说众多深度学习的基因组学应用并没有带来太多惊喜,是 "a blind man in a dark room looking for a black hat which isn't there"。在基因组学这个存在太多未知和容易迷失的领域,有效深度学习的使用需要有强大的领域知识作为支撑。DARTS工作体现了邢毅教授实验室多年以来对可变剪接机理的研究和计算方法创新的积累,由深入的领域知识和经验作为指导,是一个有效利用不同计算模型和深度学习方法的优势实现基因组学新发现的经典工作。
原文链接:
https://www.nature.com/articles/s41592-019-0351-9
制版人:子阳
参考文献
1. Baralle, F. E. & Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nature Reviews Molecular Cell Biology .18, 437-451. (2017).
2. Wahl, M. C., Will, C. L. & Luhrmann, R. The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701-718. (2009).
3. Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nature Reviews Molecular Cell Biology .18, 655-670. (2017).
4. Xing, Y., Resch, A., Lee, C. The multiassembly problem: reconstructing multiple transcript isoforms from EST fragment mixtures. Genome Research. 14(3):426-41. (2004).
5. Xing, Y., Yu, T., Wu, YN., Roy, M., Kim, J., Lee, C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Research. 34(10):3150-60. (2006).
6. Shen, S., Park, JW., Lu, ZX., Lin, L., Henry, MD., Wu, YN., Zhou, Q., Xing, Y. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proceedings of the National Academy of Sciences of the United States of America. 111(51):E5593-601. (2014).
BioArt,一心关注生命科学,只为分享更多有种、有趣、有料的信息。关注请长按上方二维码。投稿、合作、转载授权事宜请联系微信ID:bioartbusiness 或邮箱:sinobioart@bioart.com.cn。原创内容,未经授权,禁止转载到其它平台。
以上是关于原核生物蛋白质翻译的起始过程?的主要内容,如果未能解决你的问题,请参考以下文章