R语言层次聚类算法及可视化
Posted PLANTOMIX
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言层次聚类算法及可视化相关的知识,希望对你有一定的参考价值。




本节需要用到的 R 包:
# 加载包
library(dplyr) # 数据预处理
library(ggplot2) # 数据可视化
library(cluster) # 实现聚类算法
library(factoextra) # 聚类结果可视化
构建测试数据集(数据来自 R 包 AmesHousing):
# 数据预处理
ames_scale <- AmesHousing::make_ames() %>%
select_if(is.numeric) %>% # 选择数据类型为数字
select(-Sale_Price) %>% # 去除特定的数据
mutate_all(as.double) %>% # 强制转换类型
scale() # 中心化标准化数据
层次聚类算法
层次聚类算法可以分成两种:
1. 聚集聚类(Agglomerative clustering),通常叫 AGNES,理解成从个体到整体的聚类方式;
2. 分裂层次聚类(Divisive hierarchical clustering),通常叫 DIANA,理解成从整体到个体的扩散。
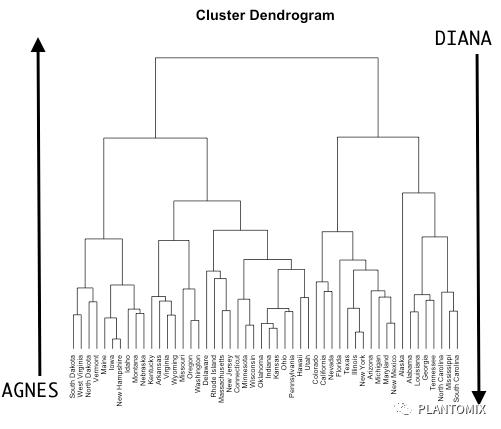
从图上可以看出两者是相反的过程。
1. Maximum or complete linkage clustering:计算 cluster1 和 cluster2 中两两元素之间的相异性,选择最大值作为距离 cluster1 和 cluster2 的相异值,这种算法更倾向于产生更加紧凑的 cluster;
2. Minimum or single linkage clustering:分别计算 cluster1 和 cluster2 内的元素的相异性,求均值作为 cluster1 和 cluster2 的相异值,这种算法通常生成一个更长的树状图;
3. Centroid linkage clustering:计算 cluster1 和 cluster2 的 “重心” 的相异值;
4. Ward’s minimum variance method:最小化集群内的总方差,每一步都将簇间距离最小的一对簇进行合并。
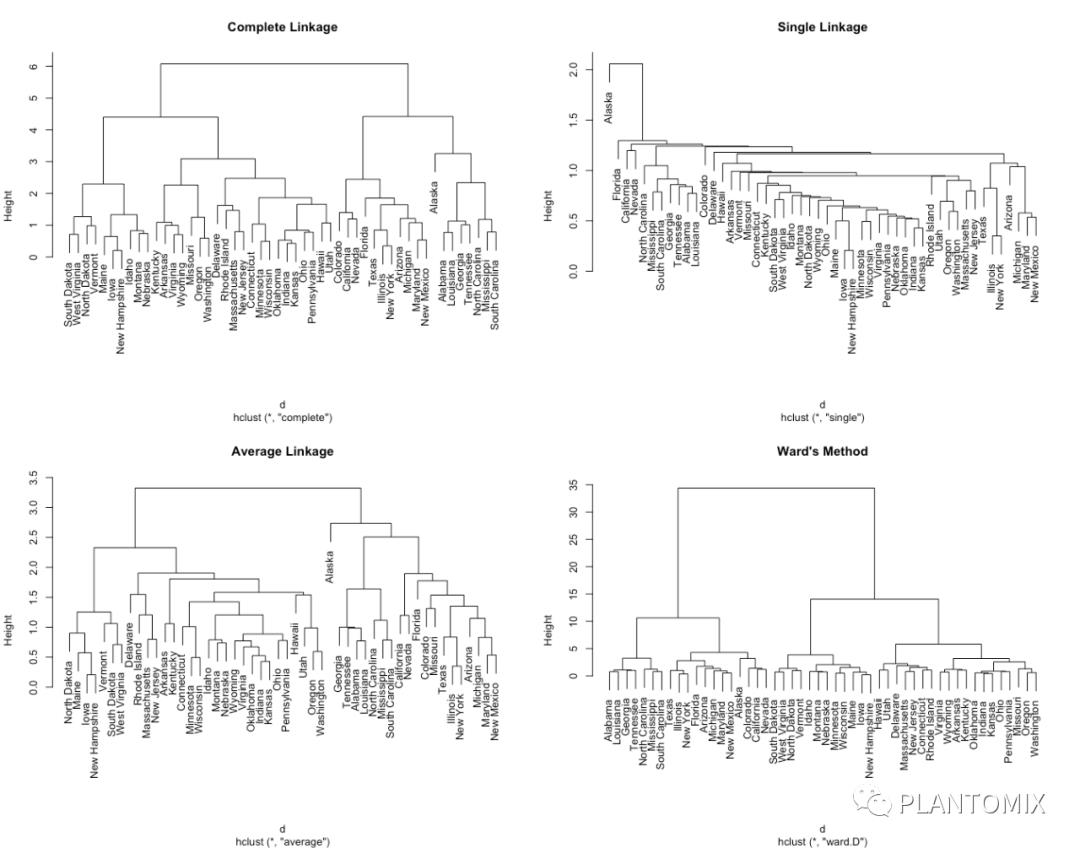
还有多其他的聚类算法,但是上面这 4 种是最常见使用最广泛的。
# 设置随机数种子
set.seed(123)
# 计算距离
d <- dist(ames_scale, method = "euclidean")
# 聚类
par(mfrow = c(2,2))
# for 循环批量计算聚类并绘图
for (i in c("complete","average","single","ward.D")) {
hc1 <- hclust(d, method = i)
plot(hc1, cex = 0.6, hang = -1,main = i)
}
由于样本有点多,图并不是很好看,但是可以看出差异。
# 设置随机数种子
set.seed(123)
agnes.ac = NULL
# 批量计算AC值
# 方法选择
m <- c( "average", "single", "complete", "ward")
names(m) <- c( "average", "single", "complete", "ward")
# 构建函数提取AC
ac <- function(x) {
agnes(ames_scale, method = x)$ac
}
# 查看AC值
purrr::map_dbl(m, ac)
# 结果
average single complete ward
0.9139303 0.8712890 0.9267750 0.9766577 这种聚类算法的函数是 diana,没有方法可以进行选择。
# 计算 divisive hierarchical clustering
hc4 <- diana(ames_scale)
# Divise coefficient
hc4$dc
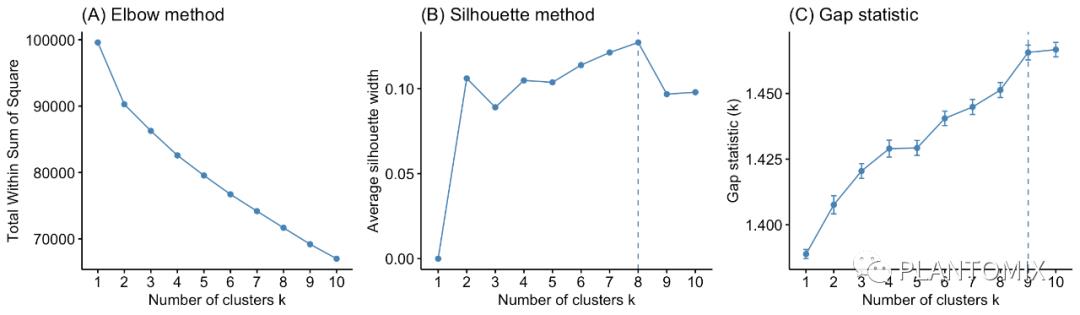
## [1] 0.9191094虽然层次聚类能够展示所有样本之间的关系,但我们还是可以像 K- 均值聚类那样看看聚成多少类比较合适。
# Plot cluster results
p1 <- fviz_nbclust(ames_scale, FUN = hcut, method = "wss",
k.max = 10) +
ggtitle("(A) Elbow method")
p2 <- fviz_nbclust(ames_scale, FUN = hcut, method = "silhouette",
k.max = 10) +
ggtitle("(B) Silhouette method")
p3 <- fviz_nbclust(ames_scale, FUN = hcut, method = "gap_stat",
k.max = 10) +
ggtitle("(C) Gap statistic")
# Display plots side by side
gridExtra::grid.arrange(p1, p2, p3, nrow = 1)
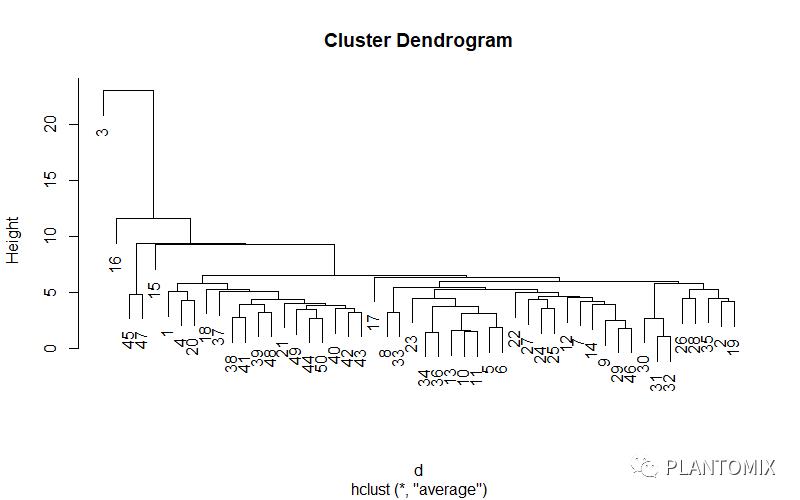
# 树状图可视化
# 选择前100个样本作为示例
d <- dist(ames_scale[1:50,], method = "euclidean")
hc <- hclust(d, method = 'average')
# 直接绘图
plot(hc)
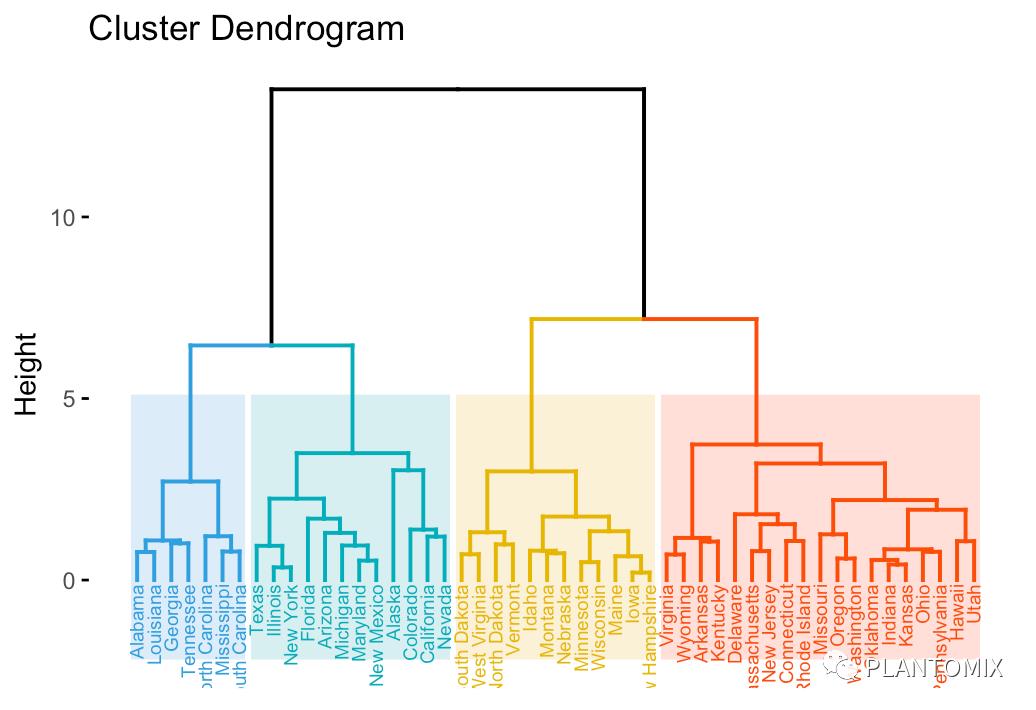
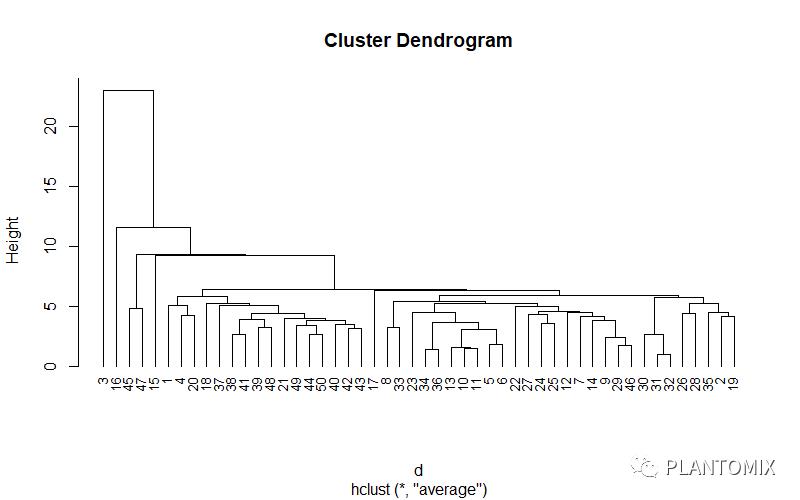
让所有样品名称排在同样的高度:
plot(hc, hang = -1, cex = 0.8)
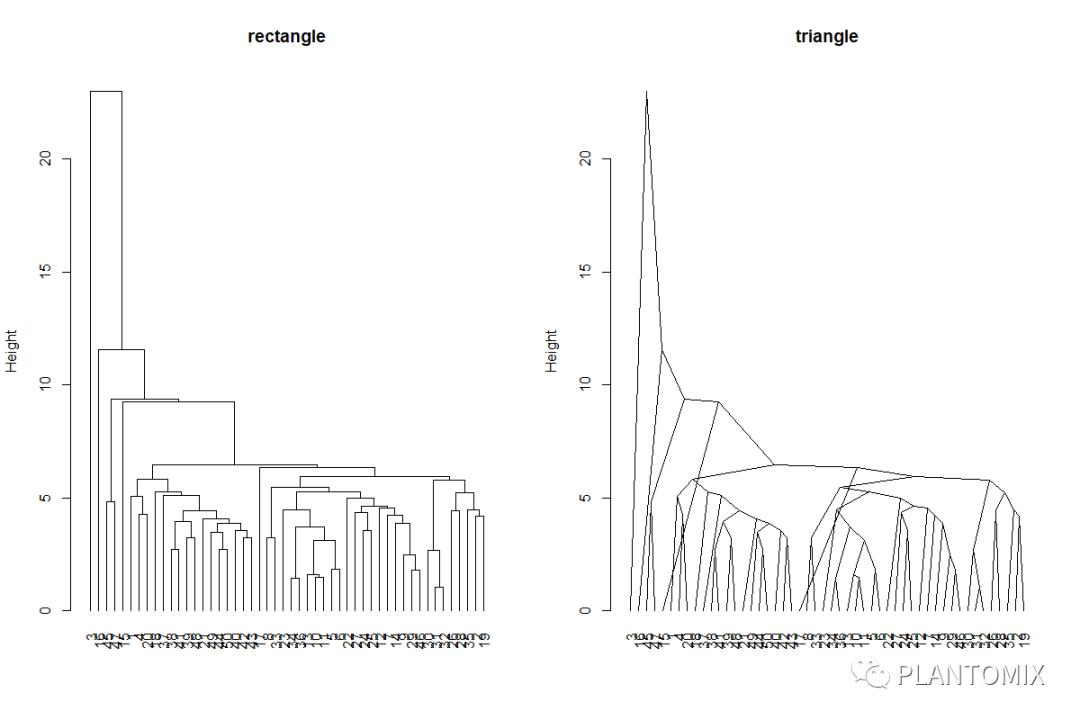
使用 plot.dendrogram() 函数对树状图进行展示需要先将聚类的结果转换成 dendrogram:
# 格式转换
hcd <- as.dendrogram(hc)
绘制两种不同形状的图:
# 两种不同形状
par(mfrow = c(1,2))
# Default plot
plot(hcd, type = "rectangle",
ylab = "Height",main = 'rectangle')
# Triangle plot
plot(hcd, type = "triangle",
ylab = "Height",main = 'triangle')
dev.off()
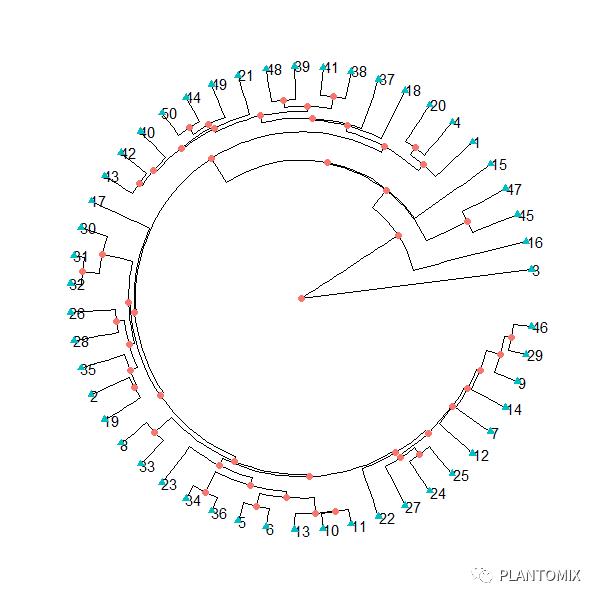
Y 叔的 ggtree 可以说是树状图(进化树)可视化的神级包了:
# ggtree
hc.ggtree = as.phylo(hc)
ggtree(hc.ggtree, layout = 'fan')+
geom_tiplab()+
geom_point(aes(shape=isTip, color=isTip), size=2)
iTOL(https://itol.embl.de/)是一个树状图可视化的神器,聚类的树状图也可以用 iTOL 进行可视化。
# 先将聚类结果转换成可用的数据
write.tree(as.phylo(hc), file = 'test.nwk')
然后将导出的文件导入到 iTOL 网站就可以了,写点配置文件就可以了。配置文件的写法可以参考:
http://ynaulx.cn/2020/01/09/iTOL%E4%BF%AE%E9%A5%B0%E8%BF%9B%E5%8C%96%E6%A0%91/#more
以上是关于R语言层次聚类算法及可视化的主要内容,如果未能解决你的问题,请参考以下文章