linux基础常用命令
Posted 生信小猪
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了linux基础常用命令相关的知识,希望对你有一定的参考价值。
cat test.txt|awk '{print NF}' #统计字符串的个数cat test.txt|awk '{print NR}' #统计每一行的行号cat test.txt|awk '{print length}' #统计每一行的字符串长度
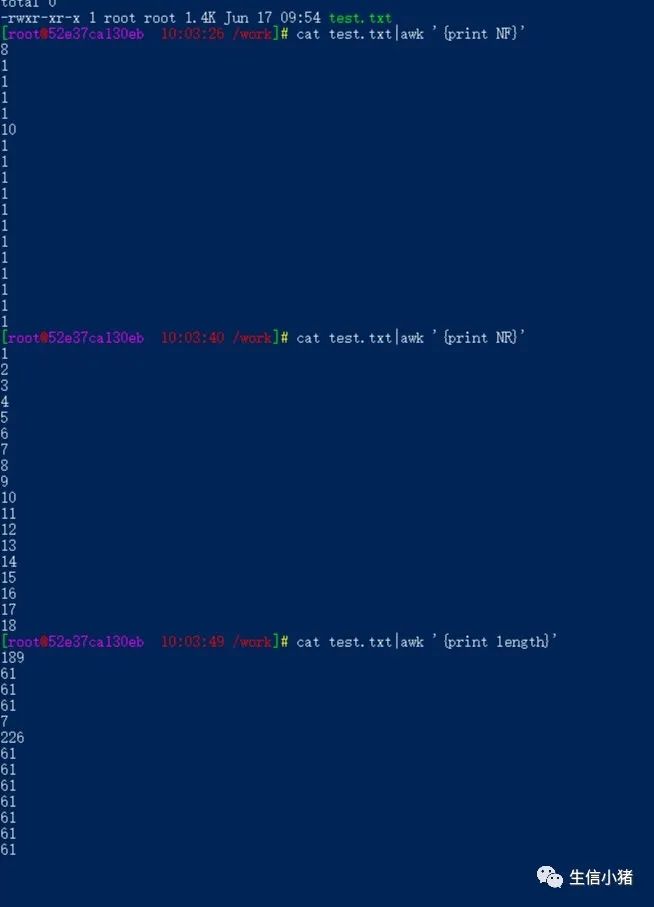
cat test.txt|awk 'NR%2==1' #删除test.txt文件中的所有偶数行cat -n test.txt #标示文件的行号tac test.txt #从最后一行开始反向查看一个文件的内容
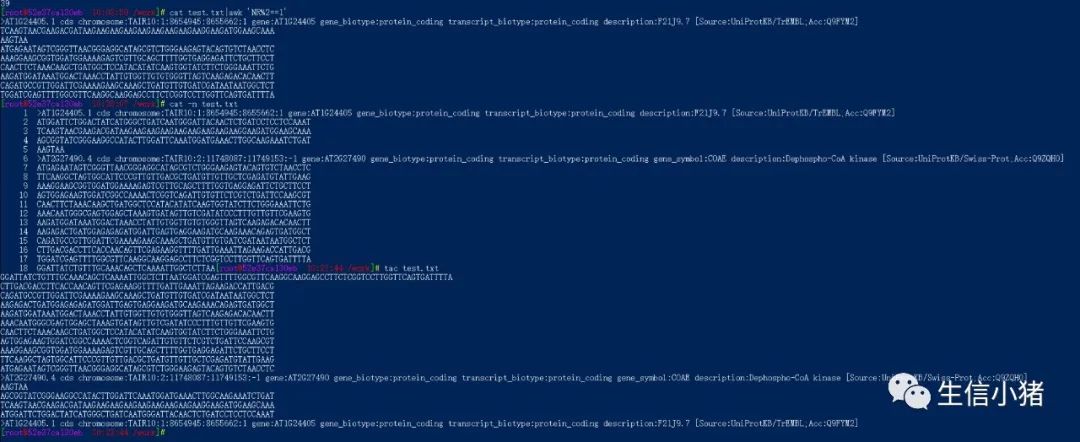
head -6 test.txt|grep '>' #查看前行中带有>号的行head -6 test.txt|grep -v '>' #查看前6行中不带>号的行wc -l test.txt #统计test.txt文件的行数wc -w test.txt #统计test.txt文件的单词数wc -c test.txt #统计test.txt文件的字节数
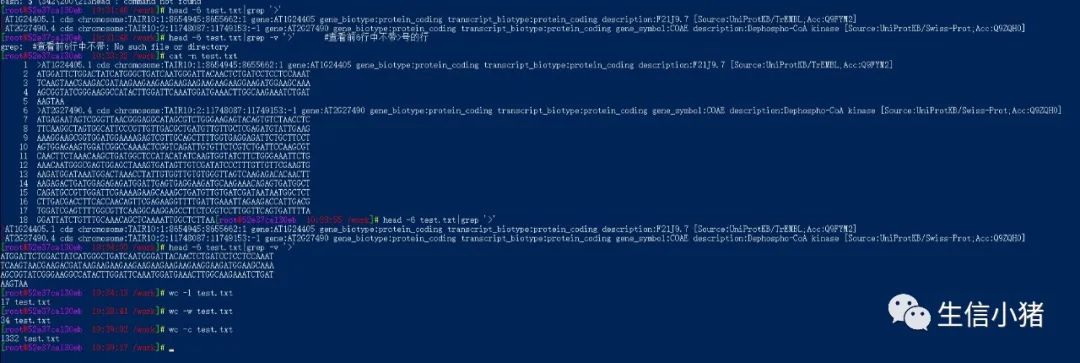
sort A.txt B.txt|uniq #取出两 A.txt 和B.txt两个文件的并集(重复的行只保留一份)sort A.txt B.txt|uniq -d #取出两 A.txt 和B.txt两个文件的交集(只留下同时存在两个文件的行)sort A.txt B.txt|uniq -u #删除两 A.txt 和B.txt两个文件的交集,留下其他的行
sort -k 3 test.txt #将文件按照test.txt以第三列来进行排序sort -k 1 test.txt|cut -f 1 #按照第1列字母顺序进行排序sort -n -k 1 test.txt|cut -f 1 #按照第1列数值大小进行排序,从小到大sort -n -r -k 1 test.txt|cut -f 1 #按照第1列数值大小进行排序,从大到小sort -k 3 test.txt|cut -f 3|uniq #去除重复行sort -k 3 test.txt|cut -f 3|uniq -c #在前面加上每一行出现的次数
awk '{print $1}' test.txt #将test.txt文件的第一列输出到屏幕awk '$3="gene" {print $0}' test.txt #将test.txt第三列是基因的列的一整行输出到屏幕中awk '$1=="Pt" && $4>=1531 && $5<=1548 {print $0}' test.txt #想要筛选染色体某一块特定的区域内的序列awk '$1=="Pt" && $4>=1531 && $5<=1548 {print $1" "$5}' test.txt #根据筛选条件,筛选出来第1、5列
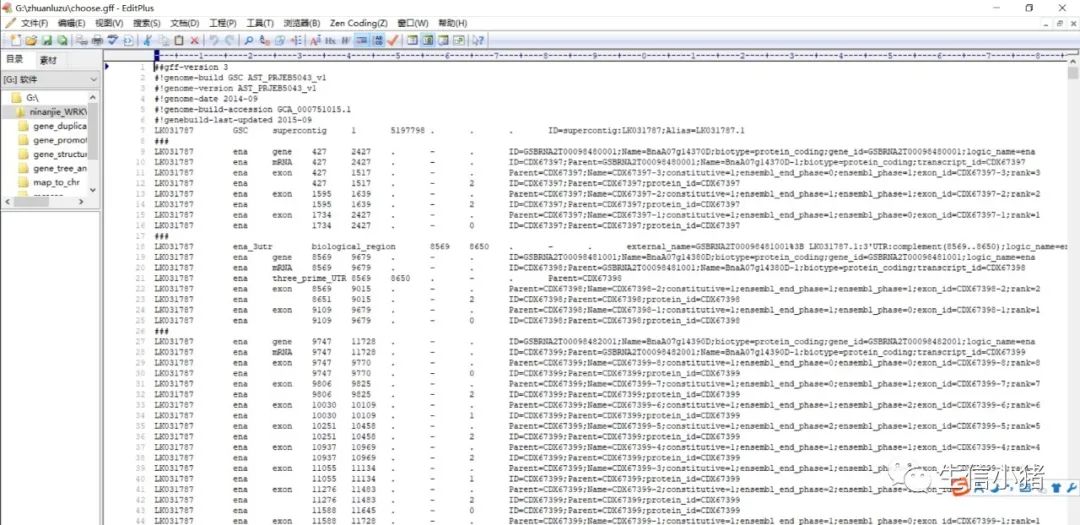
awk '$3=="gene" {print $1" "$3" "$4" "$5}' choose.gff>choose1.gff下面是生成的choose1.gff文件
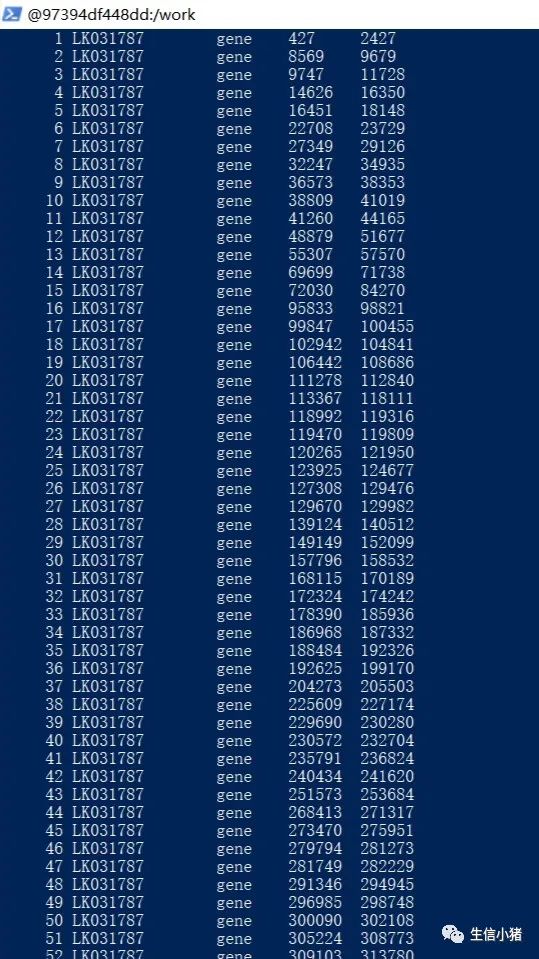
awk '$3=="exon" {print $1" "$3" "$4" "$5}' choose.gff>choose2.gff下面是生成的choose2.gff文件
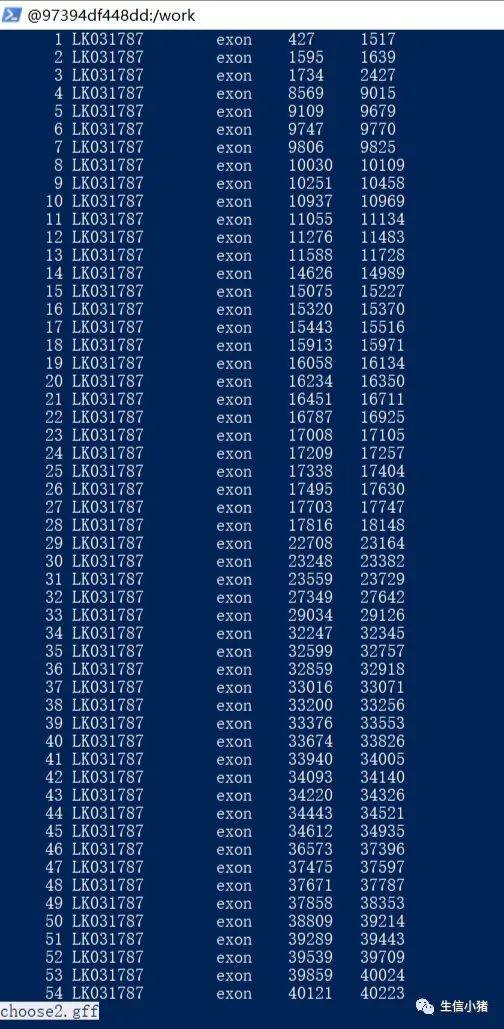
paste choose1.gff choose2.gff>choose3.gff #合并choose1.gff choose2.gff两个文件并输出到choose3中.gff
paste -d '+' choose1.gff choose2.gff>choose4.gff #合并choose1.gff choose2.gff两个文件,并用+区分,然后输出到choose4.gff文件中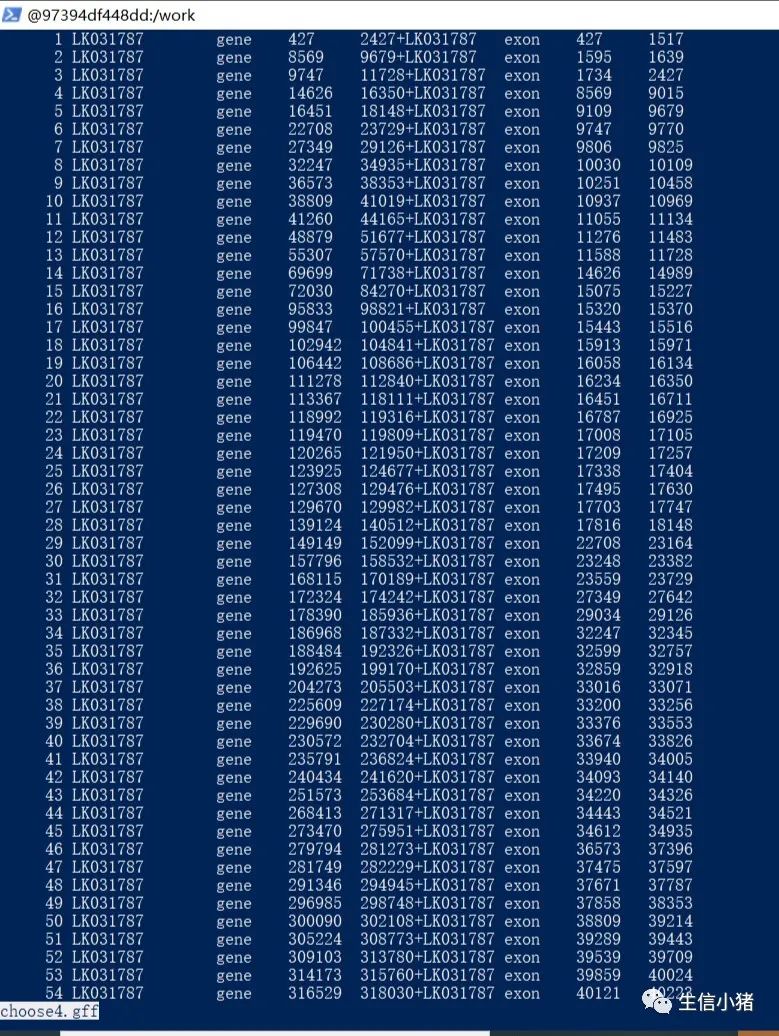
cat choose1.gff choose2.gff>choose6.gff #将choose1.gff choose2.gff两个文件进行合并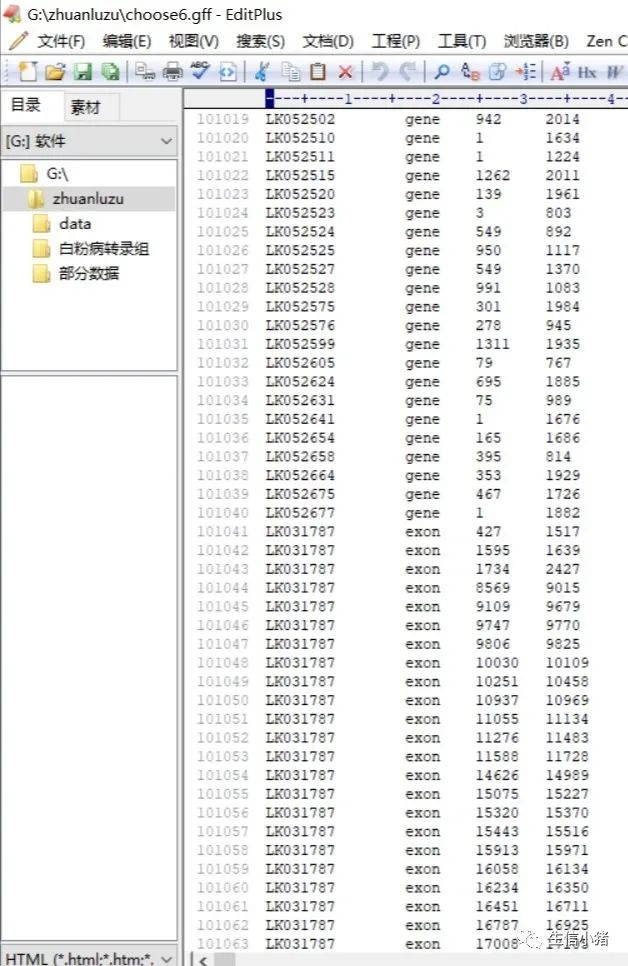
sed '/*#/d;/^$/d' choose.gff> choose5.gff #删除文件中的所有注释和空白行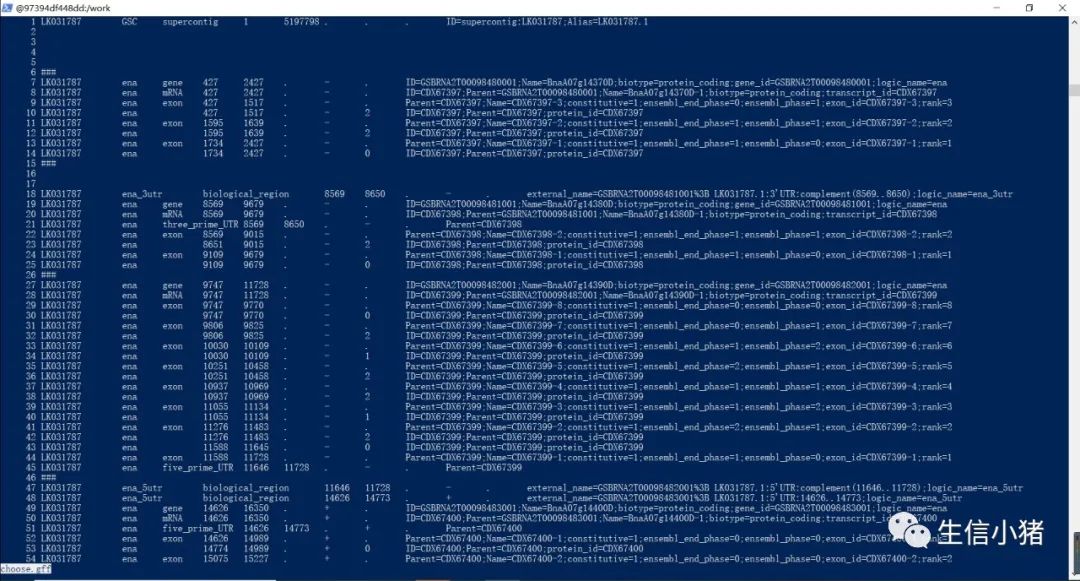
下面是生成的choose5.gff文件
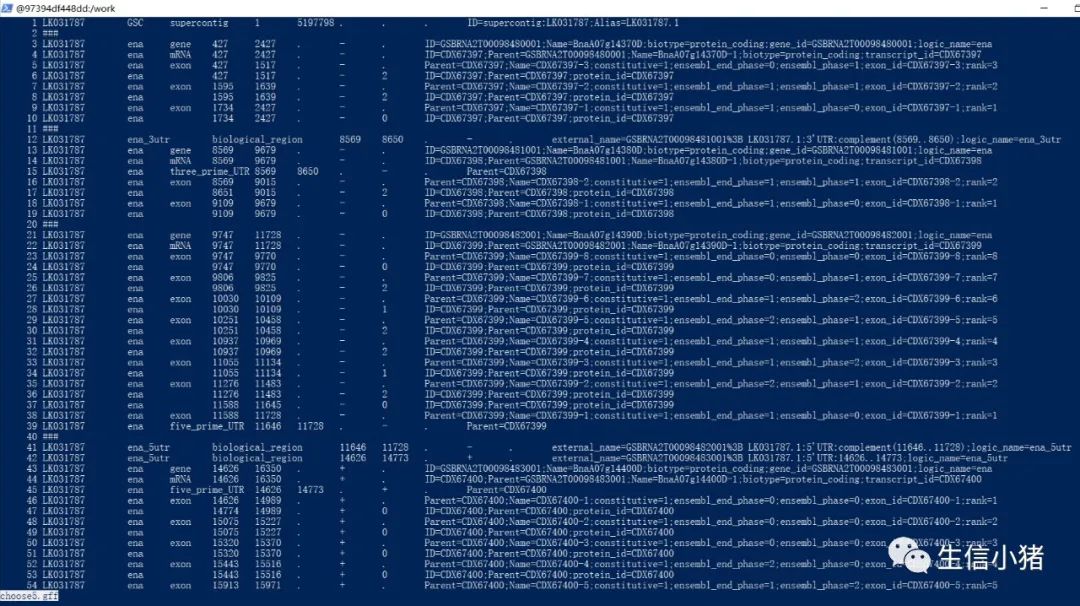
grep 'Biological Process:' S1_S2_S3_vs_R1_R2_R3.annotation.txt>result.txt #查看S1_S2_S3_vs_R1_R2_R3.annotation.txt中,包含Biological Process:的词汇,并输出到result.txtcomm -1 file1 file2 #比较两个文件的内容只删除file1所包含的内容comm -2 file1 file2 #比较两个文件的内容只删除file2所包含的内容comm -3 file1 file2 #比较两个文件的内容只删除两个文件共有的部分
grep '>' sequence.fasta|wc -l #查看含有> 的序列行号cut -d -f 3 choose4.gff #系统把choose4.gff 文件的第三列输出到屏幕当中,由于cut的默认分隔符是Tab键,而choose4.gff文件分隔符也是Tab文件分隔符也是Tab键,所以这里可以不用-d来指定分隔符sed -i 's/exon/EN/g' choose2.gff #将choose2.gff文件里的所有exon换成EN
grep [0-9] choose.gff >choose7.gff #将文件choose.gff文件中所有包含数字的行输出到choose7.gff以上是关于linux基础常用命令的主要内容,如果未能解决你的问题,请参考以下文章