八篇文章带你了解材料结构数据可视化方法
Posted 材料人
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了八篇文章带你了解材料结构数据可视化方法相关的知识,希望对你有一定的参考价值。
在材料科学中,我们对某一材料的表征(如电镜图、元素分布、带隙等)会衍生出大量的数据。在“大数据”的背景下,如何有效利用这类含有丰富内容的数据集,是基于高通量计算探索材料的主要问题和挑战。目前,创造数据驱动和自动化框架用于数据的可视化和分析是许多研究人员追求的目标。关键的问题是如何构建容易理解和操作的数据低维表达方式。
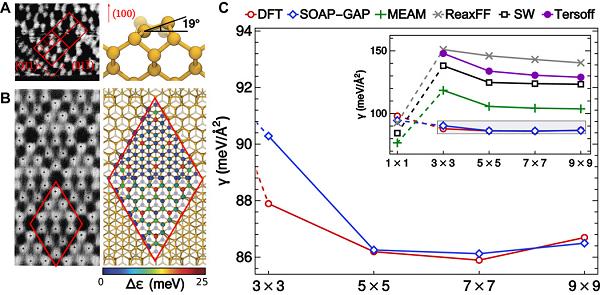
一般来说,分子或者材料的几何构象是高于三维的。然而为了对结构数据进行可视化,研究人员需要对数据进行低维化,即将结构转化成点,使其能够在纸面或者屏幕上进行直观体现。用于蛋白质骨架结构表征的Ramachandran图就是一个经典的例子,利用这种二维图基于仅仅两个扭转角(torsion angles)就可以对具有66个几何自由度的多肽分子进行可视化。实现低维嵌入的关键是如何定义原子结构中距离尺度。机器学习(machine learning)是目前用来描述预测原子结构的主要方法之一。例如研究人员在晶胞里建立了基于分子动力学构象数据集的SOAP(smooth overlap of atomic positions)-GAP(Gaussian approximation potential)模型1。基于这种模型,研究人员能够准确描述在宽泛的温度和压力范围内,块体和缺陷材料的性质。如图1所示,STM图像表征了在重构硅表面的二聚体具有倾斜角(tilt angle)。而基于SOAP-GAP预测,并结合密度泛函理论(DFT)发现,这一弛豫结构来源于Jahn-Teller变形,且倾斜角的角度经过模型预测为19度左右。
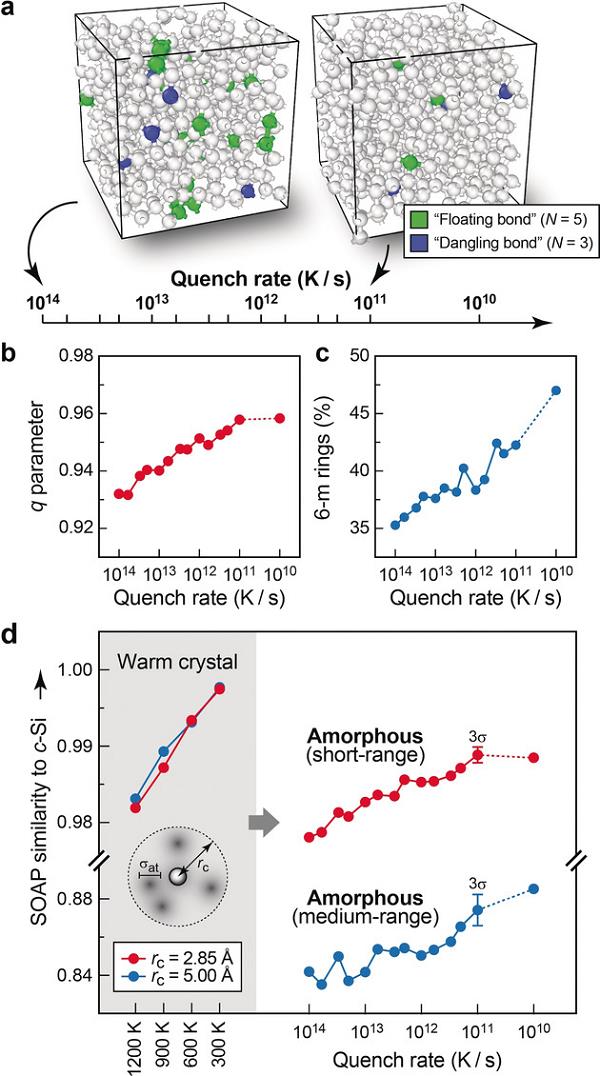
实现材料及分子结构的自动比较和绘图首先需要描述原子环境(atomic environment)。而所谓的原子环境则是指以特定原子为中心,一定半径范围内的原子(化学物种及位置)所构成的。在机器学习中,一个优秀的原子环境描述符(descriptor)应该不随同一物种原子的转变、旋转、重排等不会改变物理性质的操作而一同改变。因为非恒量的描述符通常基于原子的共价链接,具有低维属性,会丧失许多几何信息。而恒量描述符则可以尽可能地保留原子环境和结构的几何信息,由此再通过维度还原的低维嵌入以达到绘制低维结构图谱的目的。例如Deringer等2人基于机器学习研究了非晶硅的原子结构量化可行性。研究人员利用局部稳定性量化描述了原子最近邻和次近邻结构,并利用一分析方式分析了不同淬灭速率下取向度不同的非晶硅网络。此外,这一方法还将非晶硅的配位缺陷和稳定区域联系在一起,并追踪液态硅在玻璃化过程中的能量过渡状态(图2)。
图2 基于机器学习模拟非晶硅网络的熔融-淬灭过程2
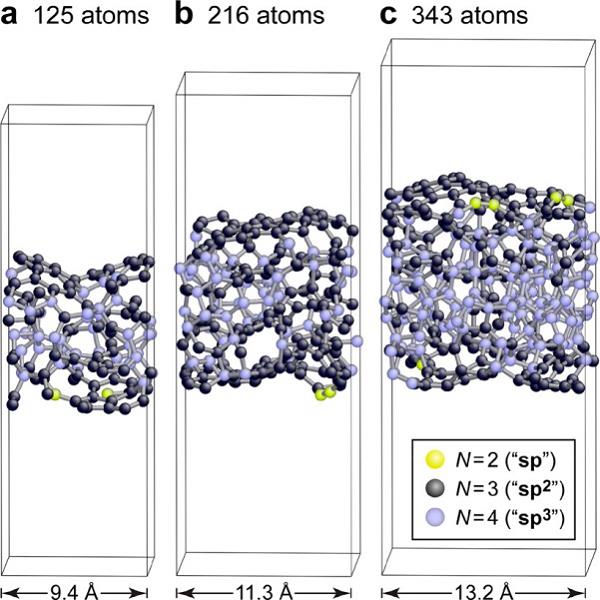
凭借其优异的力学性能,四面体非晶碳在涂层领域拥有广泛的应用;同时四面体非晶碳也能够作为理想的电极材料用以检测生物大分子。然而,尽管应用范围广泛,四面体非晶碳表面具有复杂的原子尺度结构和化学反应活性,至今还未被深入理解。剑桥大学的Deringer等3人将机器学习、密度泛函紧束缚(tight binding)(DFTB)以及DFT结合起来用以研究这一长期困扰的问题。研究人员基于第一性原理建立了一系列四面体非晶碳表面的原子论模型,以局部结构指纹作为特征,能够在不同的系统尺度中提供一系列结构。再利用蒙特卡洛算法和由DFTB得到的相互作用,可以研究四面体非晶碳的逐步氢化过程(图3);而利用DFT-机器学习则可以研究原子级的氧化机制。这一尝试为理解非晶材料表面提供了新的可能,还能以空前的准确性来研究表面结构的化学功能化。
图3 基于机器学习模拟的四面体非晶碳表面的典型结构模型3
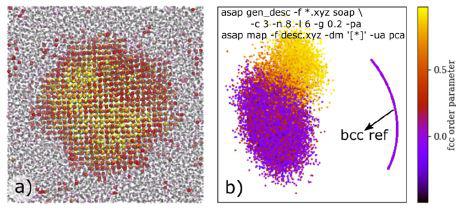
在晶体形成过程中,也可以利用自动绘图技术来理解结构(例如研究成核过程的结构异质性情况)。一般来说,材料的固化都来源于小晶体的成核。而尽管模型研究不少,但是对面心立方晶核表面是否存在体心立方取向一直具有争议。这一争议主要是因为体心立方的物理界限不好判断,同时常用的局部键取向参数也无法区别体心立方和界面原子。而Cheng4等人则开发了新型策略可将宏观理论和仿真计算有机结合在一起,以研究晶体成核。为了实现这一目的,研究人员基于多簇模型建立用以研究的理想原子论体系。紧接着发展了一种与多簇模型一致的热力学框架,要求更少的假设,并适用于分子动力学或者蒙特卡洛研究。如图4所示的主元分析(PCA)图,是基于在冷却液体中的固体核内的原子环境的SOAP描述符绘制的。利用经典的面心立方取向参数,根据他们与面心立方的相似度可对环境进行着色。由图3可知,分别代表面心立方和类液体模块的数据点揭示了核中心和液体之间存在平滑渐变的过渡状态,并且不存在明显的代表体心立方局部取向的额外密度峰;相反,作为参比的体心立方环境则与目标数据点之间存在明显的分隔现象。这一方法为“成核过程中不存在体心立方取向”提供了颇具说服力的证据
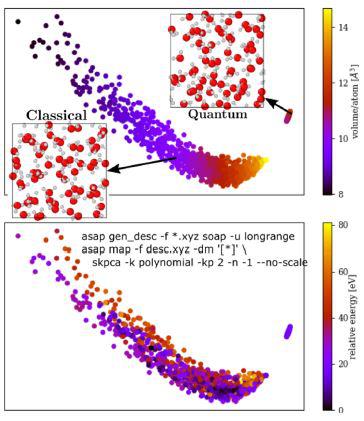
计算物理化学领域的一个中心命题是基于量子力学的基本法则利用第一性原理来预测材料的性质。然而,这类方法的计算成本高,阻碍了在有限的温度里阻碍对诸如热容、密度以及化学势等进行准确预测。针对这一问题,Cheng5等人将先进的自由能方法和数据驱动的机器学习原子间势能结合起来,用以实现更加严谨可靠的预测方式。研究人员基于DFT,并考虑了量子核运动、不和谐波动以及质子失序等因素,实现了对液态水的热力学性质预测。利用经典分子动力学模拟,研究人员收集了1000个液态水结构;而基于这些数据又进一步提取了经过几何优化的数据——基于氢核的量子力学本质实现对液态水结构的量子力学化。由图四可知,经典和量子的水形式分隔成了两簇;其中经典水构象在能量和摩尔体积两个方向均有宽泛的分布,揭示了核-量子扰动在调节不同水相热力学稳定性中扮演的角色。
图5 1000个经典液态水构象和593个量子力学液态水结构的数据绘图5
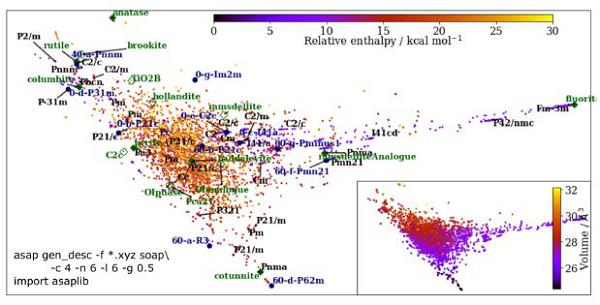
对晶体多晶相的相稳定性进行研究和预测一直是计算材料科学的重要课题。然而这类研究一直极具挑战。最主要的原因是这类研究首先要求势能最小化,并执行复杂的自由能计算以对有限温度的熵效应进行解释。而Reinhardt6等人则发展了一种框架能够通过获取晶体结构来实现相稳定性预测和研究。研究将随机结构研究(RSS)和机器学习有机结合在一起用以研究固相的自由能,并以此实现对未知相的热力学相行为研究。研究人员选择多晶相众多的二氧化钛作为研究对象,收集上千个截然不同的原子配位、晶胞形态等结构数据制成数据集,如图6所示形成了包含4690个局部稳定结构的结构相似度图谱。在这一图中,材料的性质如焓或者单元晶胞体积都可以一目了然,再通过映射分析已知相和未知相就可以快速识别研究中出现的相数据。
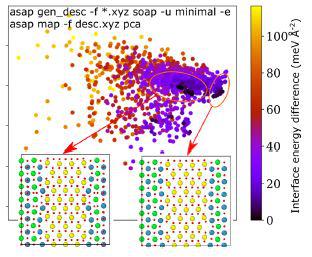
结构研究也可以扩展到界面系统中,用以揭示其他途径难以获取的稳定构型。由于界面的存在打破了晶体对称性,对界面结构进行数据分析更加具有挑战。图7显示了钛酸锶(STO)和钐掺杂二氧化铈(110)/(110)界面结构7。PCA图谱能够自动识别界面能量,每个点均代表了局部能量最小值的构型,而相对能量则被用作颜色标尺,与横轴具有强关联关系。钐掺杂二氧化铈具有萤石结构的离子导体,而STO则是具有钙钛矿结构的绝缘体。从PCA图中可以识别两种簇:分别是与块体相简单连接形成的理想界面结构相似的结构数据,以及包含重构结构的数据群组。
图7 钛酸锶和钐掺杂二氧化铈(110)/(110)界面结构的PCA图7
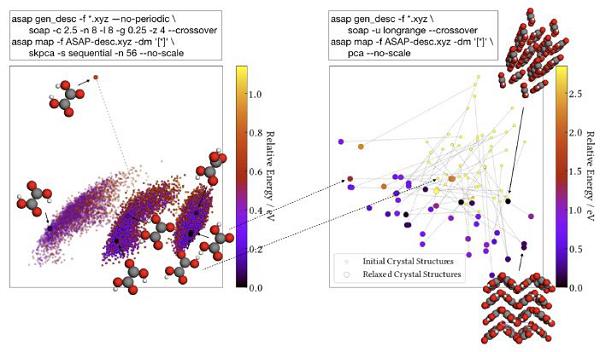
主元分析也可以用于鉴别组分固定体系的多晶中的不同之处。以草酸为例,在图8中8,左图是展示了气相中,草酸在势能表面的七种稳定构象异构体(conformer)结构,并且根据质子取向以及纵轴将这些结构进行排列。而在右图中,基于主元分析和随机结构研究揭示了草酸的48个块状晶体结构,初始的随机结构与相应的弛豫构象利用灰线进行连接,并且随机和优化的晶体结构处在不同的区域。几乎所有的优化结构都是分隔清楚的,表明晶体具有许多稳定的最小值,这与气相截然不同。图8的左右两图强调了分子结构的不同层面,其中左图基于质子取向显示了分子内层面;而右图则揭示了分子间相互作用的不同之处。因此,这两图的结合可以为更深入理解分子晶体形成中的结构参数提供了新的模式和机会。
图8 基于主元分析和机器学习揭示了草酸分子晶体结构8
1. Machine learning unifies the modeling of materials and molecules. Sci. Adv., 2017, 3, 12, e1701816.
2. Quantifying Chemical Structure and Machine‐Learned Atomic Energies in Amorphous and Liquid Silicon. Angew. Chem. Int. Ed., 2019, 58, 7057-7061.
3. Computational Surface Chemistry of Tetrahedral Amorphous Carbon by Combining Machine Learning and Density Functional Theory. Chem. Mater., 2018, 30, 21, 7438-7445.
4. Bridging the gap between atomistic andmacroscopic models of homogeneous nucleation. J. Chem. Phys. 2017,146, 034106.
5. Abinitio thermodynamics of liquid and solid water. Proc. Natl. Acad. Sci.U. S. A. 2019, 116, 1110−1115.
6. Predicting the phase diagram of titanium dioxide with random search and pattern recognition. Phys. Chem. Chem. Phys., 2020,22, 12697-12705.
7. Determining interface structures in vertically aligned nanocomposite films. APL Materials 2019, 7, 061105.
8. Mapping Materials and Molecules. Acc. Chem. Res. DOI: 10.1021/acs.accounts.0c00403.
未经允许不得转载,授权事宜请联系kefu@cailiaoren.com。
历经四年,材料人网已经汇聚了300余人的专业计算科技顾问团队,他们来自于中国、欧洲、日本、澳洲等全球知名大学的博士、博后、青椒、教授,专业性强、技能可靠,可以承接各种难度、各个方面的计算模拟工作。

客服微信二维码
以上是关于八篇文章带你了解材料结构数据可视化方法的主要内容,如果未能解决你的问题,请参考以下文章
数据结构和算法难理解?可视化动画带你轻松入门!
防劝退!数据结构和算法难理解?可视化动画带你轻松透彻理解!
用Python爬取分析某东618畅销商品销量数据,带你看看大家都喜欢买什么!
数据之美笔记
Echarts数据可视化一个项目带你学会酷炫的数据可视化
Python入门可视化:22个完整数据可视化小例子,带你玩转可视化~