正则表达式与grep
Posted 生信媛
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了正则表达式与grep相关的知识,希望对你有一定的参考价值。
正则表达式(regular expression, regex)是一个重要且实用的概念,我时常提起却从未细谈。一怕能力不够说不清楚反而会误导人,二是已经有无数前人撰文介绍。考虑日常用到的 grep, sed, awk里经常需要用到正则表达式,于是开一个小系列,介绍如何在 grep, sed, awk中适用正则。
正则表达式(regular expression)的概念,最初来自于20世纪40年代的两位神经学家(Warren McCulloch, Walter Pitts)研究神经元时提出的想法。后来数学家Stephen Kleene在代数学中正式描述了这种被他称之为“正则集合”的模型。并且,他还发明了一套简洁的方法表示正则集合,也就是正则表达式。
目前最快速的文本搜索工具 grep就内置了正则表达式。 grep起源于Unix中的ed编辑器的一条命令 g/RegularExpression/p, 读作“Global Reular Expression Print”,即运用正则表达式的全局输出。由于这个功能太过实用,于是就从ed中独立出来,演变成了 grep以及扩展版本的 egrep。都知道grep因为有正则表达式所以很强大,但是正则表达式为何如此强大呢?
正则表达式的强大之处在于它是一套语法,分为两个部分,元字符(metacharacters)和普通文本字符(normal text characters)。
以语言类比,“我爱正则表达式”这句话可以抽象成“主谓宾”结构,主语是"我",谓语是"爱",宾语是“正则表达式”。这种结构还适用于其他语言,比如说英语就是"I love regular expression". 这种语法结构就是元字符,而构成句子的语言就是普通文字字符。
正则表达式有很多流派,不同流派之间的差异在于对元字符的支持程度。以下的元字符适用于GNU版本的grep, sed, awk. mac自带的是BSD版本。
匹配单个字符的元字符:
| 元字符 | 匹配对象 |
|---|---|
| . | 匹配单个任意字符 |
| [...] | 匹配单个列出的字符 |
| [^...] | 匹配单个未列出的字符 |
| \char | 转义元字符成普通字符 |
提供技术功能的元字符
| 元字符 | 匹配对象 |
|---|---|
| ? | 匹配0或1次 |
| * | 匹配0到n次 |
| + | 至少一次,最多不限 |
| {min,max} | 至少min次, 最多max次 |
匹配位置的元字符
| 元字符 | 匹配对象 |
|---|---|
| ^ | 匹配一行的开头 |
| $ | 匹配一行的结尾 |
其他元字符
| 元字符 | 匹配对象 |
|---|---|
| 竖线 | 匹配任意分割的表达式 |
| (...) | 限定多选结构的范围,标注量词的作用范围,为反向引用捕获元素 |
| \1, \2 | 反向引用元素 |
小字:awk, grep, sed支持perl的正则系统,而perl的正则表达式是及其强大的,后续可能会说到。
grep的强大之处它所做的事情就只有在文本搜索”正则表达式“定义的模式(pattern),如果找到就打印出来。可以使用 man egrep查看所支持的参数。
egrep [options] pattern [file]
egrep [options] [-e pattern]... [-f FILE]... [FILE...]
# 参数参数
-e PATTERN: 定义多个模式
-f FILE: 从文本中读取模式
-w: 匹配整个单词
-v: 反向匹配
-i: 忽略大小写
-x: 仅仅选择整行匹配结果
-c: 计数
-n: 输出表明行号
-A/-B NUM: 同时输出后/前几行
注: grep有基础和扩展两个模式,基础模式支持的元字符较少,而 egrep表示扩展的grep,支持的元字符较多。
检查测序结果的接头
在分析的质控阶段,需要检查给出的结果是否含有接头(adapter)。除了fastqc, 还能用grep检测是否有接头序列。
你可以去illumina的官网根据公司测序的protocol查找对应的接头: https://support.illumina.com/downloads/illumina-customer-sequence-letter.html
或者是直接看FASTQC的配置文件中检测的接头
# 下载测试数据
fastq-dump -X 10000 SRR1553606 --split-files
然后用部分的接头对fastq文件进行搜索
egrep -B1 'TCGGAA' SRR1553606_1.fastq
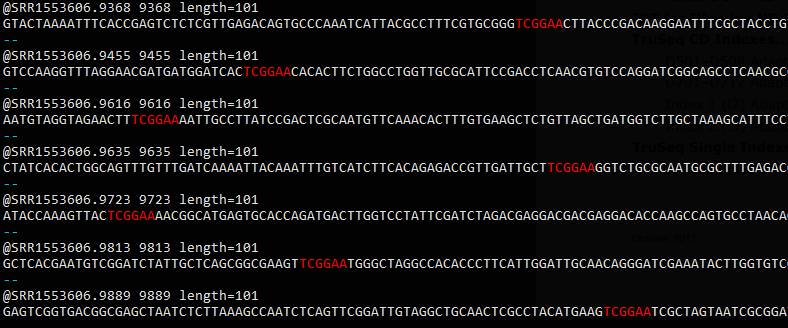
被找到的序列并非出现最开头,你可能希望是开头有几个其他碱基,然后跟着接头序列.
egrep -B1 '^[ATGC]{0,5}TCGGAA' SRR1553606_1.fastq
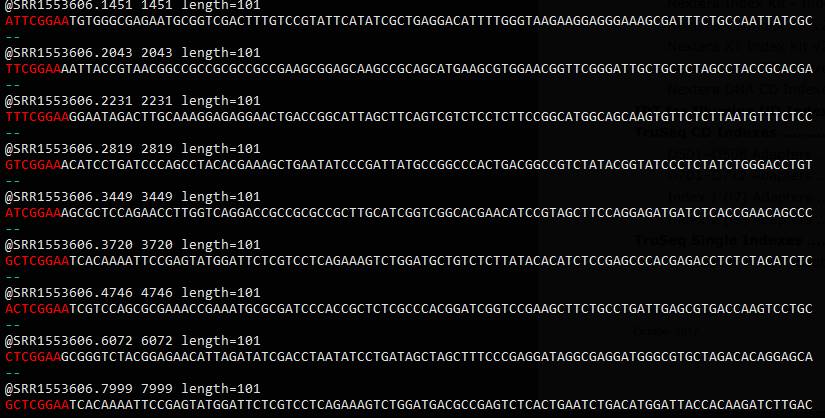
基因搜索
一般而言,大家都回去TAIR上查找基因的区间。但是实际上我们可以先下载好拟南芥参考基因组及其注释文件,然后直接用正则表达式确定区间,用 bedtools提取序列进行了。
# 下载序列
wget -c -4 -q http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_chromosome_files/TAIR10_chr_all.fas
# 下载GFF文件
wget -c -4 -q http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/TAIR10_gff3/TAIR10_GFF3_genes.gff
# 基因的信息
wget http://www.arabidopsis.org/download_files/Genes/TAIR10_genome_release/gene_description_20131231.txt.gz
在注释文件中查找某一个基因如SPL9, 并获取他的位置信息.
grep -i -w 'spl9' gene_description_20131231.txt
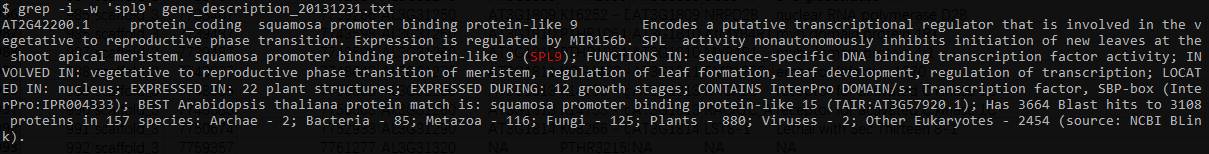
grep -e 'AT2G42200.1' TAIR10_GFF3_genes.gff | grep 'mRNA'

当然以上命令可以连成管道, 并且和bedtools合用,就可用直接获取DNA序列。
bedtools getfasta -fi TAIR10.fa -bed \
<(grep -i -w 'spl9' gene_description_20131231.txt | \
cut -f 1 | xargs -i grep -i {} TAIR10_GFF3_genes.gff | grep mRNA | cut -f '1,4,5')
你可以将其定义成一个函数,存放在配置文件中, 随后就能进行调用该函数。
这样子就不需要去TAIR才能找基因序列了,而且速度更快,没有网络延迟哦
by 徐洲更
by 徐洲更
by 徐洲更
以上是关于正则表达式与grep的主要内容,如果未能解决你的问题,请参考以下文章