2 相关技术及原理
Posted gamedev˚
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了2 相关技术及原理相关的知识,希望对你有一定的参考价值。
2 相关技术及原理
2.1 Hadoop相关技术和原理
本节主要对基于Hadoop平台的相关技术,如:HDFS、YARN和MapReduce三大模块进行原理介绍 ,为后续基因测序在Hadoop平台上的搭建作相应的准备。
2.1.1 HDFS分布式存储系统
Hadoop分布式存储系统,即Hadoop Dirstributed FileSystem,简称HDFS1。由于Hadoop集群的基础架构设施,使得HDFS是一个能提供可扩展性和高可靠性的基于Java的文件系统,并且适用于大型商业服务器集群或是廉价服务器上,从而可以提供PB级的存储容量和上千个服务器的计算能力。
此外,HDFS还是一个高容错的分布式存储系统,理论上可以无限扩展集群的存储能力,并与YARN服务(主要负责Hadoop集群中的资源管理和任务调度工作)协调各种并发任务调度。通过在多台服务器节点之间分配存储资源和计算资源,使得存储资源可以随需求线性增长,并协调各个服务器之间的计算能力。
HDFS有以下特点来确保数据高效存储在Hadoop集群中,并具有高可用性:
1. 机架感知意识,在Hadoop集群中客户端进行数据访问时,会自动考虑节点的物理位置,将离作业节点最近的数据块传输过去。
2. 保证最小的数据转移,Hadoop将计算任务移动到数据所在的节点上,这样能保证处理任务发生在数据所在的物理节点,从而显著减少机架之间的网络传输的I/O流。
3. 动态诊断HDFS存储系统的运行状况,并重新平衡不同节点上的数据。
4. 回滚操作,HDFS支持对某一状态的文件系统进行快照保存,这样允许操作员在升级后恢复以前的HDFS版本,以防止发生人为或系统错误。
在高可用的集群中,在两台独立的机器分别配置NameNode。在任何时候,只有一个NameNode处理Active状态,另一个处理Standby状态,在必要时候可以提供快速故障转移。HDFS的联邦政策,允许在一个集群中部署多个子NameNode,可以扩展文件块的镜像文件的大小,并管理联合集群。
在Unix系统架构中,每次磁盘都有默认的数据块,文件都以块的形式存储的,同样,在HDFS也有块(block)的概念,但是比Unix的文件系统块大得多,默认为128MB。HDFS的架构图如图2-1所示:

图2-1 HDFS架构图
对分布式文件系统中的块进行HDFS抽象可以使得一个文件的大小可以大于网络中任意一个磁盘的容量。从本地文件系统将一个文件复制到HDFS,该命令调度Hadoop文件系统的fs命令,并提供一系列子命令,包括-copyFromLocal。在下面的命令中,本地文件local.txt会被上传到HDFS中,路径为/user/elon/remote.txt。
% hadoop fs -copyFromLocal local.txt hdfs://localhost/user/elon/remote.txt2.1.2 MapReduce并行计算框架与Yarn资源调度器
Hadoop MapReduce2是一个计算框架,在进行MapReduce编程时,要编写用于数据处理的map函数和reduce函数,并以可靠并容错的方式在数千个节点的大型群集的大型商业硬件上并行处理大量数据。
通常,计算节点和存储节点是相同的,允许框架在数据已经存在的节点上有效地调度任务,达到数据本地化的效果,从而在整个集群中具有非常高的执行效率。
在Hadoop2.0以后,Hadoop社区将资源管理和任务调度的功能从原来的MapReduce中分离出来,形成现在的YARN3,Apache Hadoop YARN架构 如图2-2所示:
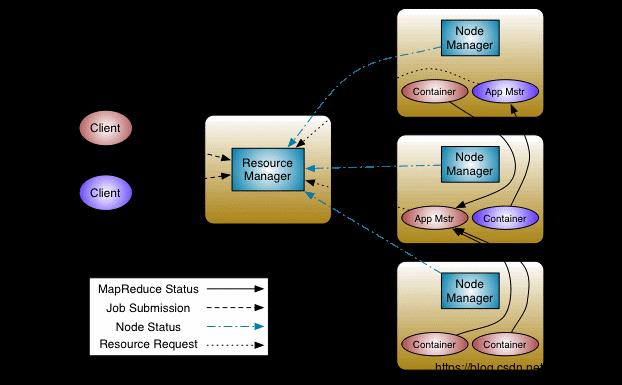
图2 2 Apache Hadoop YARN架构图
从上面的YARN的架构图可以得知,YARN[13]是由资源管理器和作业调度及各个节点的监控所组成的,包含一个全局的资源管理器(ResourceManager,简称RM)和每个应用程序的管理器(ApplicationMaster,简称AM),其中应用程序可以是单个的作业或者是一个DAG(有向无环图)的作业流水线。
MapReduce程序就是在YARN上进行资源分配和作业调度的,集群中的每个计算节点的主机都运行着一个节点管理器(NodeManager,简称NM),负责监视机器上各个容器的资源私使用情况,而作业是部署在每个容器(Container)中进行计算的,对于一个大型的任务,可能会被拆分为多个map任务而被部署在各个容器中来并行计算。
对于一个作业的完整运行流程的阐述如下:首先从客户端提交作业,随后被分配到各个计算节点的container容器中,并会在其中一个计算节点生成AM任务管理器,AM了解本次任务执行所需的资源,并会向RM进行资源请求,随后RM将命令NM资源管理器向该节点中的容器分配相应的计算资源。在整个任务的计算周期内,Container会定时向AM发送自己MapReduce程序的运行进展情况,NM对自己节点上的Container容器的资源使用情况进行监控,并将其报告给RM。
2.2 全基因组测序相关技术和处理流程
在进行全基因组测序相关处理流程的设计4时,主要涉及三个大的流程步骤(如下图),全基因组的测序分析步骤如图2-3所示。
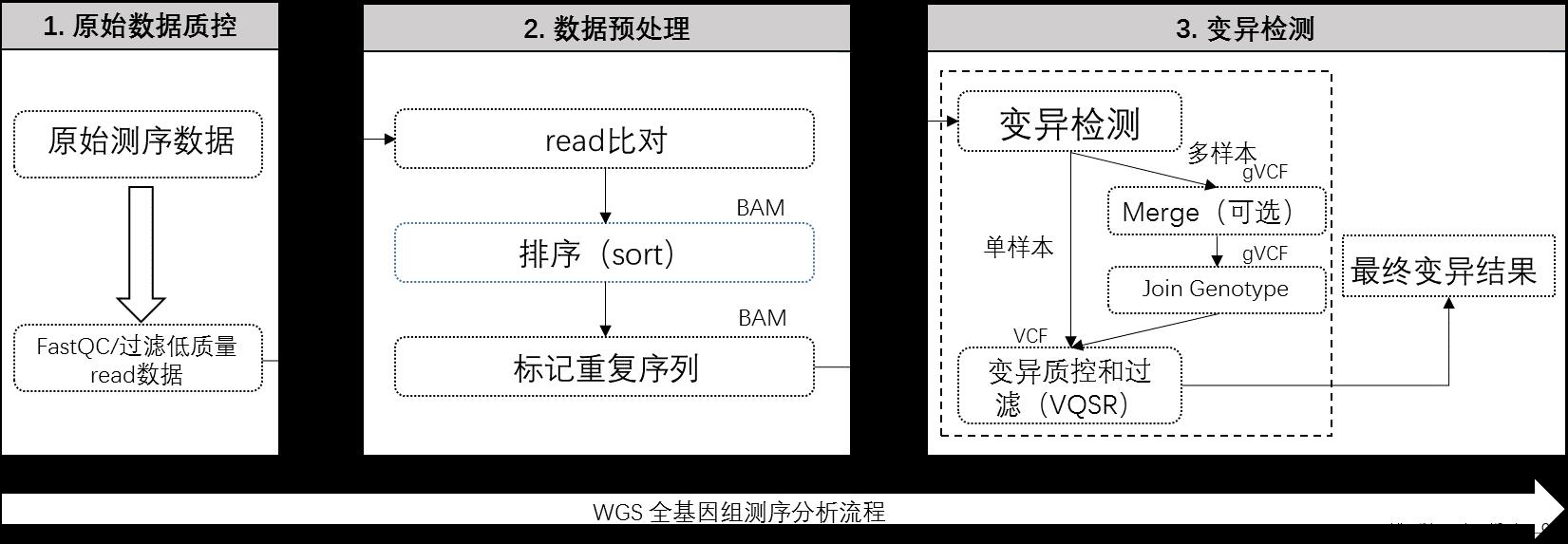
图2-3 全基因组的测序分析步骤
第一步是对测序仪中得到的原始基因组数据进行数据指控,只有在对需要进行测序的数据进行数据质控后并保证了数据源的有效性,后面测序的结果才是真实有效的。在第二步中,对测序数据进行预处理,而这其中主要包括三个步骤:read比对、排序以及标记重复序列,并对标记的重复序列建立索引,最终作为变异检测的输入数据。在第三步中,进行主要的变异检测操作,而这里又分为两种方式,第一种是对单样本数据直接进行变异检测处理,直接得到变异结果。第二种是对多个样本组数据进行分批变异检测处理,得到分离的gVCF文件,最后再合并成一个VCF变异文件,得到最终的基因变异集合。
2.2.1 原始数据质控
在全基因组测序研究中,以illumina为首的测序流程大多数都是运用边合成边测序的技术,其中需要经历一系列的化学反应。在这个合成的过程中随着化学合成链的增长,DNA聚合酶的活性会不断下降。另外,由于测序仪在刚开始化学合成反应时不够稳定,同样会带来碱基质量值的波动。
测序数据的质量好坏会直接影响我们的基因测序的下游分析和最终的变异检测的结果。因此我们需要在进行下游分析之前进行数据的质量控制检测5。但不同的测序平台其稳定性和测序的准确性也有差异,目前在数据指控方面使用较多的是FastQC这款软件。
FastQC6是一个Java程序,能够比较高效地来计算,并能以html的格式给出read的质量值和碱基测序错误率,形成最终的基因样本数据的测序报告。在给出的报告中,会包含几个重要的指标并以图谱的形式展现出来,指明原始数据可能还存在着哪些问题,并通过其质控结果作出相应的后续工作,比如是直接在后续分析流程中使用、或是进行部分切除后使用,亦或是将该原始数据直接丢弃不使用等。
在本次研究所使用的大肠杆菌的全基因组的测序样本中,对所有的样本都进行了数据质控的操作,并根据质控报告做了相应的处理。下面我选取SRR3226036_1号样本 进行数据质控的流程展示,基本的测序数据信息如图2-4所示。
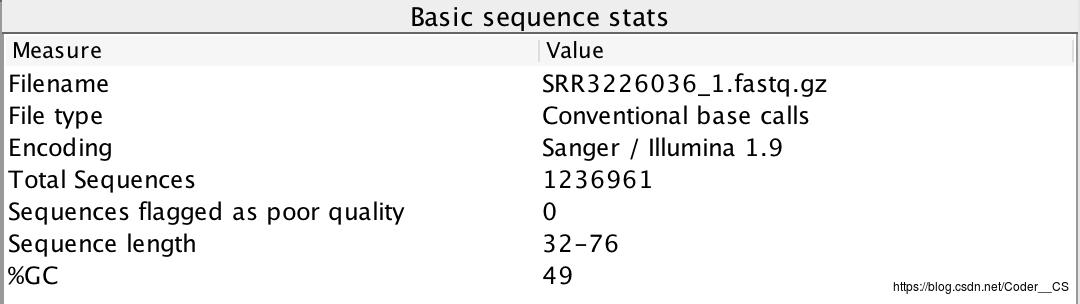
图2-4 基本的测序数据信息
从上图中,我们不难得到该SRR3226036_1号样本数据的基本信息,其中就包括测序数据的文件名、文件类型,测序仪的编码类型等基本信息,后面还给出来本次质控的总测序read数是123万多条,测序read中被标记为低质量的测序读长为0,每条测序read的长度在32-76之间。
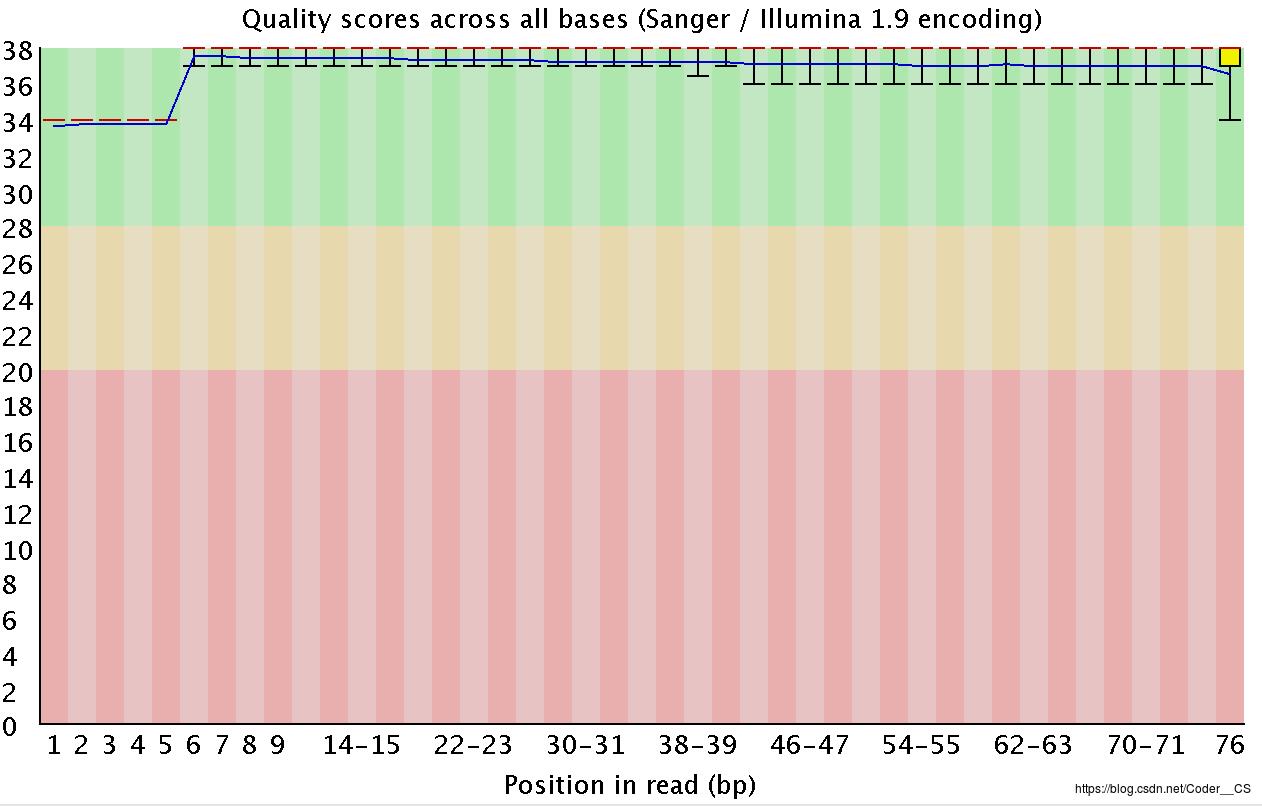
图2-5 read的碱基质量值分布图
read的碱基质量值分布如图2-5所示,展示的是该样本质控中read的碱基质量值分布图,其中横轴是read上碱基的位置分布,纵轴是碱基质量值。在该图中碱基质量值都大于35,而且各个read位置的碱基值波动很小,可以看到图中质量值的分布都在绿色背景(代表高质量)的区域,说明这个样本的碱基质量很稳定,这其实是一个非常高质量的样本质控结果。
各读长中碱基的含量占比如图2-6所示,展示的是该测序样本中各个碱基的含量占比情况,我们可以发现在读长为小于20以及读长大于75的区域,其碱基含量分布是波动较厉害的,说明在这两个区域的化学合成反应是不稳定所导致的,也就是在反应的开始阶段测序仪内部环境还处于不稳定的状态,以及在化学反应后期生物酶已逐渐失活所引起的,恰好应证了在本节最开始处提及的一般测序read不会太长的情况,这也是在数据质控的基本信息那张图片所展示的测序读长在32~76之间的缘故。因为只有这一块的碱基含量占比是稳定的,而且各个碱基之间的占比差距不大,表明这部分读长的数据是相对可靠的,适合拿来做基因测序的下游分析。
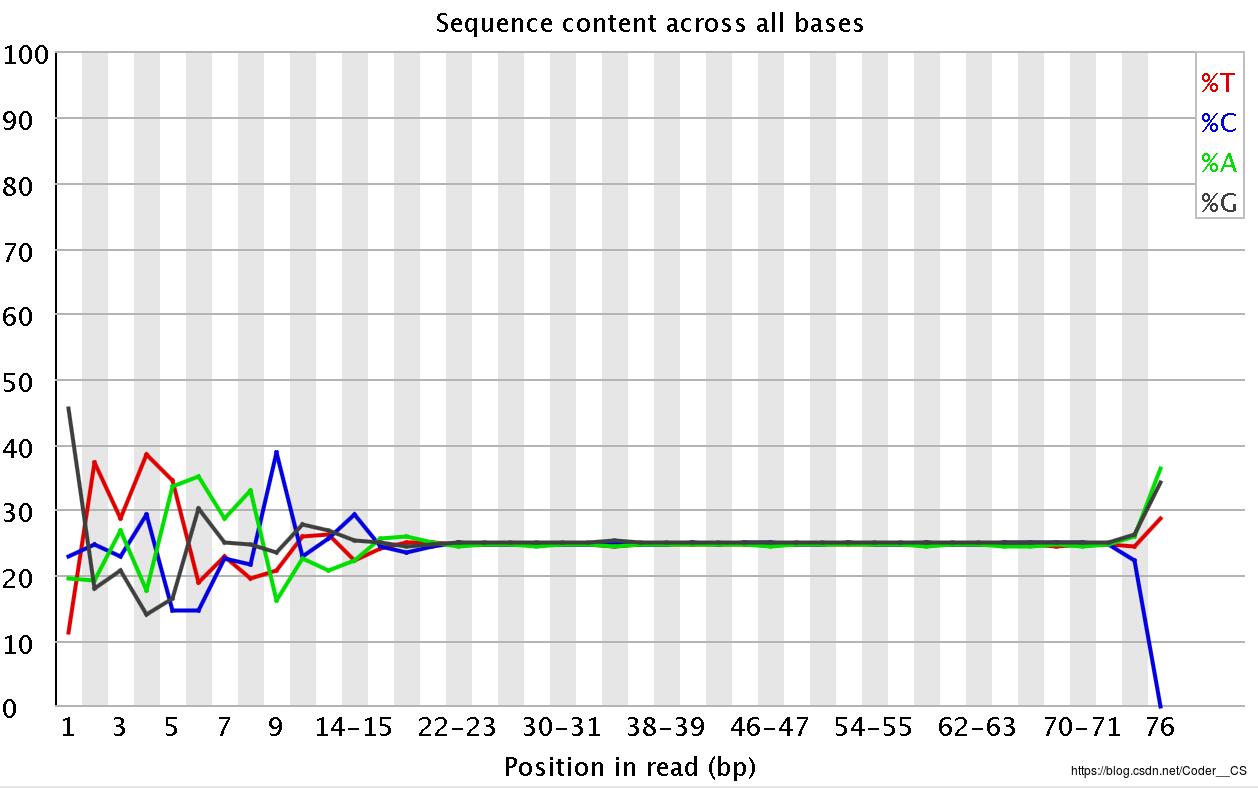
图2-6 各读长中碱基的含量占比图
这一节主要对测序样本数据进行质量分析控制,将不适合进行下游分析的数据样本切除或丢弃。下一节会详细介绍基因测序处理流程的各个分析阶段,展现如何从一组样本数据得到最终的变异检测文件。
2.2.2 数据预处理
(1) 第一步:序列比对
全基因组测序的短序列(read)存储于FASTQ文件里面,在我的测序样本数据中是用fastq.gz的压缩格式表示的,这样的gz格式文件,在序列比对时也是被支持的。它会先进行解压再开始比对。虽然这些短序列在测序仪中进行化学反应时是在一起的,原本都来自于有序的基因组库中,但在经过化学合成和碱基提取之后,原本的read序列的前后顺序关系就已经不存在了。所以,FASTQ文件中紧挨着的两条read之间都是随机来自于原本基因组样本中某个位置的短序列而已。
因此,我们需要先把这一大堆的短序列捋顺,将这些短序列与该物种的参考基因组 作比对,通过一个read一个read的比对,找到每一个read在参考基因组上的位置,并在文件中标记好每个读长的位点信息,从而得到比对后的数据文件,这个过程就称为测序数据的序列比对。
测序数据的序列比对本质上是一个按照参考基因组来寻找最大公共子字符串的过程。而目前用的比较广泛的是BWA 软件,它将使用BWA-MEM算法[17]进行序列比对。下面是BWA的官方文档[18]说明,其中指出了用于序列比对的API:
Usage: bwa mem [options] <idxbase> <in1.fq> [in2.fq]
其中第一个参数[option]是可选参数,包括程序处理时所分配的线程数,最小的seed长度以及read的类型等信息。第二个参数是指代参考基因组的BW索引文件。第三个和第四个参数指代的是输入测序数据,包括R1.fastq和R2.fastq文件。输出数据包含各read序列的位点信息,可以采用重定向的方式输出到其他文件中。具体的示例如下:
在上面的示例中,需要补充的是samtools软件的使用,这个在下面排序阶段会有提及,这里对其进行简单说明,最后将比对的文件进行重定向到管道中,并交由samtools的view方法进行自动检测数据格式,并最终输出BAM格式的文件并保存在SRR3226036.bam中。
(2) 第二步:排序
由于在上一步中只是将原始短序列读长与参考基因组进行序列比对,并记录各个序列的位点信息随即就输出了,而对原始序列未进行移动,它们还是随机分布于BAM文件中的。在这一步排序中,就是通过第一步中保存的各个序列的位点信息,需要将比对记录按照顺序从小到大排序下来,才能进行后续的去重复等操作。
这里,我们将会用到另外一款流行的基因测序的软件Samtools 。这里通过其官方API文档对Samtools的一个简单介绍。samtools一共有五个模块的功能,其中包括indexing(建立索引)、editing(对读长序列进行处理,包括替换BAM头、去除重复以及修复元数据信息等)、file operations(对测序样本文件操作,包括切分、合并、排序以及转换文件格式等)、stats(读取文件的状态信息等,包括BAM索引信息、计算测序深度以及生成状态信息等)和viewing(对BAM标记进行解释,以及将SAM、BAM和CRAM三种格式进行转换)。在本次测序流程中,主要使用这款软件完成排序和建立索引的操作,因此还会再后面的建立索引的流程中使用到它。在Samtools[19]官方文档中,比对命令如下:
Usage: samtools sort [options...] [in.bam]
其中第一个参数[options…]是可选参数,在本次排序处理中,会用到的有-@ 整数,设置用于排序和压缩的线程数;-m 整数,设置分配给每个线程的最大内存资源;-O 格式类型,指定输出的格式类型,这里包括sam、bam和cram三种格式,我们需要的是bam格式,用于下一步的标记重复操作;-o 文件,指定最终输出到一个文件中保存,而不是将其直接输出到屏幕上的这种标准输出形式;以及-T PREFIX,指定临时文件的文件命名中的首字母,最后这些临时文件会在排序结束后删除。第二个参数[in.bam]指代的就是已包含各个测序序列的参考位点信息的比对文件。具体的示例如下:
$SAMTOOLS/samtools sort \\
-@ 4 \\
-m 4G \\
-O bam \\
-o SRR3226036.sorted.bam \\
SRR3226036.bam \\
-T PREFIX.bam \\
&& echo "*** BAM文件排序完成"
(3) 第三步:标记重复序列
在基因测序的处理流程中,我们通过化学反应得到DNA液,并放到测序仪中将其测序并切分为每个短读长序列,分离成每个碱基对的形式,得到我们所需的原始基因组数据。这样一系列操作需要通过物理(超声)打断或者化学试剂(酶切)的方式来打断原始的DNA序列,其中就存在DNA的降解的问题,再加上有些基因数据在原本的基因组中含量就相对稀少,会存在局部DNA浓度过低的问题,因此需保证每种DNA片段在原来的基因组中的相对含量。
因此,我们需要选择适合实验的特定长度范围的序列来进行PCR扩增,并加入到测序仪中测序保证数据的完整性问题。而基因组中重复序列的来源,实际上就是PCR扩增过程所导致的。PCR扩增本质上就是通过化学反应的方式将原始的某一段DNA序列复制多次。
但是要注意的是,PCR技术是为了增大微量的DNA片段的浓度含量,但由于整个化学反应都在同一个试管中进行,因此其他一些密度并不低的DNA片段同时也会被放大好多倍,这时再取样去测序仪上测序的时候,这些浓度并不低的DNA片段就很可能会被重复取到相同的多条,并通过测序仪中的测序得到样本数据。因此我们需要对上一步中有重复的读长数据进行标记或者去除操作。
事实上,目前用的比较多的工具有Samtools和Picard,它们都可以去除重复序列,只是一个是直接删除重复序列,另一个可以仅标记但可保留重复序列,在最后的变异检测期间将其忽略。下面分别对这两种去除重复的方式进行介绍:
第一中,Samtools的方式:
Usage: samtools rmdup [-sS] <input.srt.bam> <output.bam>
在这个API中,可选参数-s指代的是仅对SE读长序列去除重复序列,-S指代的是对待PE读长和SE读长一样,都进行去除重复序列操作;第二个参数
java -jar picard.jar MarkDuplicates \\
I= SRR3226036.sorted.bam \\
O= SRR3226036.sorted.markdup.bam \\
M= SRR3226036.sorted.markup_metrics.txt \\
&& echo "*** 对BAM文件进行重复标记"
这里总共需要三个参数,第一个是输入上一步已排序好的数据源BAM文件,第二个是输出的带有重复序列位点信息标记的BAM文件,第三个是对重复序列的位点信息进行标记的文件。这里还可以在第一个参数前使用REMOVE_DUPLICATE=true或REMOVE_SEQUENCING_DUPLICATES=true选项,表示在此操作中重复序列片段会从原BAM文件中直接删除掉。
这一步完成之后,我们需要为重复序列比对阶段得到的文件创建索引。创建索引的好处是它能够让我们可以随机访问这个文件中的任意位置并提供灵活性。通过在前面提及的Samtools软件来建立索引,命令如下:
Usage: samtools index [-bc] [-m INT] <in.bam> [out.index]
其中,第一个可选参数[-bc]分别是对BAI-format和CSI-format的序列格式转换为BAM格式的文件,第二个参数-m 整数是为CSI指数设置最小的区间大小。在我的处理测序流程中具体的示例如下:
2.2.3 变异检测
到目前为止,我们已经完成了基因测序的前两大模块,数据质控和数据预处理,接下来就到了对样本数据进行变异检测的阶段了。这是本次全基因组分析流程创建的最终目标——得到样本集合的变异集合,可以为后续基因组学的科研提供有力的数据支撑。
变异检测的内容一般会包括:SNPs、indels、CNV和SV等。在这次的基因变异检测流程搭建中,主要涉及的是前两个SNPs和indels的变异检测。我们采用的生物信息学的分析工具The Genome Analysis Toolkit ,简称GATK。它是由The Broad Institute公司开发的用于二代重测序数据分析的一款软件。GATK是鉴定种系DNA和RNAseq数据中SNP和indels的行业标准,也是目前行业使用率最高的基因数据变异检测工具。GATK4.0的范围现在扩大到包括体细胞短变异调用,并且能够处理拷贝数(CNV)和结构变异(SV)。除了不同的调用者本身之外,GATK还包括许多实用工具,用于执行相关任务,如高通量测序数据的处理和质量控制,并捆绑了流行的Picard工具包。
通过GATK的官方API文档[20]中查看,可以得知和变异检测有关的方法有两个,它们分别是UnifiedGenotyper 和HaplotypeCaller 。
UnifiedGenotyper是根据每个基因组来调用SNPs[21]和indels[22]。该工具使用贝叶斯基因型似然性模型来同时估计N个样品群体中最可能的基因型和等位基因频率,为每个样品产生一种基因型。其输入数据可以是产生多种不同调用的read数据,输出则是一行未筛选高敏感性的VCF格式的文件。对于大量的样本组文件SNP调用,其分析示例如下:
java -jar GenomeAnalysisTK.jar \\
-T UnifiedGenotyper \\
-R reference.fasta \\
-I sample1.bam [-I sample2.bam ...] \\
--dbsnp dbSNP.vcf \\
-o snps.raw.vcf \\
-stand_call_conf [50.0] \\
[-L targets.interval_list]其中,-T指代的是调用的方法类型;-R指代的是参考基因组;-I指代的是输入的样本数据,如果有多个则输入多个样本名;–dbsnp表示目前已知的某物种基因变异位点文件,一般会由大型的研究机构发布出来;-o是指代vcf格式的输出文件;-stand_call_conf 指代应该调用变体的最小phred-scaled置信度阈值,默认值是50.0;可选参数-L指代的是一个或多个要运行的基因组区间。
而对于另外一种基因变异检测的方法是通过HaplotypeCaller来实现,除了可以像UnifiedGenotyper一样,对变体的数据进行整体调用,得到变异检测的VCF文件,它和上一种方式的不同之处在于,它还可以通过局部重新组装haplotypes来调用SNPs和indels,侧重点在于对局部的样本文件进行变异检测,换句话说,只要程序遇到显示变化迹象的区域,它就会丢弃现有的映射信息,并完全重新组合该区域中的读取。
这一特性对于多样本数据的基因检测尤为有效,因为在对多样本进行变异检测时,可能刚开始的数据并不多,在测序流程进行到一部分的时候,该项目研究中需要再新增一部分新的样本数据,当使用原来UnifiedGenotyper的方式来进行变异检测生成VCF数据时,需要对全量数据重新进行分析计算。但是在这里,有了HaplotypeCaller的分析方式,就不存在全量数据的概念了。因为每个样本数据都视为整体的一部分,而对于每部分的样本变异检测都会生成gVCF文件,每个gVCF文件之间是隔离的,在有新的样本数据加入时,只需生成增量样本数据的gVCF文件即可。在我的测序处理流程中就是采用的这种先生成gVCF文件的方式。在我实际的测序流程中,具体的示例如下:
同样,这个HaplotypeCaller相关的分析命令和第一种UnifiedGenotyper方法的分析命令相似,唯一的不同之处在于新增了一项参数–emitRefConfidence GVCF,这项参数是固定的,指明本次命令变异检测的是单样本数据,产生的是gVCF文件格式。最后对于该样本组下的所有gVCF文件,通过GATK的另一个方法GenotypeGVCFs来进行合并处理,具体的使用示例如下。
java -jar GenomeAnalysisTK.jar \\
-T GenotypeGVCFs \\
-nt 4
-R reference.fasta \\
--variant SRR3226034.g.vcf \\
--variant SRR3226035.g.vcf \\
…
--variant SRR3226042.g.vcf \\
-o E_coli_K12.vcf \\
&& echo "*** 合并所有的gVCF文件为VCF文件"该方法是专门对HaplotypeCaller生成的gVCF文件进行genotyping联合,输入参数是一个或多个HaplotypeCaller gVCF进行genotype,输出参数是一个已经组合的genotyped的VCF。最终生成的E_coli_K12.vcf文件,即是E_coli_K12物种来自样本组中的全基因组变异检测结果。
2.2.4 相关文件格式概述
(1) FASTA文件格式
FASTA[23]作为存储有顺序的序列数据的文件后缀,这里的序列数据指的其实就是能表示DNA或者蛋白质的一条字符串。FASTA文件表示的序列是有顺序的,我们可以通过各个read中碱基位点序列数,就可以知道某个DNA碱基在某个基因组上的准确位置,这个位置会用所在序列的名字和所在位置来表达,比如基因数据比对的结果等。
FASTA文件主要由两个部分构成:序列头信息(有时包括一些其它的描述信息)和第二行具体的基因序列数据。头信息独占一行,以大于号(>)开头作为识别标记,包括染色体的名称信息和一些其他信息。
>NC_000913.3 Escherichia coli str. K-12 substr. MG1655, complete genome
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTGTCTGATAGCAGCTTCTGAACTG
GTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATACTTTAACCAATATAGGCATAGCGCACAGAC
AGATAAAAATTACAGAGTACACAACATCCATGAAACGCATTAGCACCACCATTACCACCACCATCACCATTACCACAGGT
AACGGTGCGGGCTGACGCGTACAGGAAACACAGAAAAAAGCCCGCACCTGACAGTGCGGGCTTTTTTTTTCGACCAAAGG
上面是一个FASTA文件的例子,这是大肠杆菌K12.coli微生物基因的部分序列。在序列头信息中标注的是这个基因文件来自NC_000913.3号染色体,来自大肠杆菌K-12的全基因组数据。
(2) FASTQ文件格式
FASTQ[24]文件格式是目前存储测序数据最普遍、最公认的一个数据格式。前面FASTA文件存储的是来自该生物已排序已知的参考基因组的序列信息,而FASTQ存储的是来自测序仪得到的原始测序数据。这些原始数据是通过测序仪生成的图像数据转换过来,也是文本文件。文件大小依照测序量(或测序深度)的不同而存在较大差异,小的可能只有几M,如微生物的基因组,而大的则有几十甚至上百G,文件后缀通常都是.fastq.fq或者.fq.gz(gz压缩格式)。
@SRR3226034.1 1/1
TCCACGTTGCGACCGACAGCGCCACACGCTTCTGGTGGCGAGGCGACAACTCATTCCAGTTCAGCACATAGCGCT
+
@<@6@=6C@@FFDGGEGGGGGGGGGGGEDGGGGG8EFFGD>FGCEGCEDEEEFGG<FFDGGAFFF,9<,C+>,
@SRR3226034.2 2/1
CCTTGCCGAGGGCCCGGCCCTCGACGCCCACGCGCCAGCCGCCGCTGGGCTGCCAGGCCAGCGCGCCGTACAGCG
+
AACCCC+@8::FGGGCGGGGGC7FEGGGGGGGFGGDGGGEGG7FF7=7C=:=E8C,DC87C+@+@F7@+B+,:+
上面是从NCBI上下载的K12.coli物种的基因组测序样本SRR3226034的部分数据,从这个例子可以看出,FASTQ数据有着自己独特的格式:每四行成为一个独立的单元,我们称之为一个读长,简称为read,这是测序的最小单位。具体格式如下:
1) 第一行:以‘@’开头,是这一条read的名字,根据测序仪测序时的状态信息转换生成的,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现;
2) 第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这是真正的DNA序列信息,其中N字符代表的是测序时那些无法被识别出来的碱基;
3) 第三行:以‘+’开头,一般会空着;
4) 第四行:用ASCII码标明测序read的质量值,表示的是每个测序碱基的可靠程度。
在后面的全基因组测序中要用到这两种数据格式,其中FASTA是参考基因组的存放格式,FASTQ是基因样本数据的存放格式。
(3) gVCF文件格式
对单个样本Pair对数据进行基因测序时,最终得到Genome Variant调用格式文件,缩写为gVCF。它是一种文本文件格式,存储变体和非变体位置的测序信息,以一种紧凑的格式表示基因组中所有位点的基因型、注释和其他信息 。
gVCF是一种轻量的变体存储格式,可以将其压缩成gzip文件(.genome.vcf.gz)。随后可以将得到的gCVF文件进行索引,创建一个.tbi格式的文件,并与现有的VCF工具(如tabix和IGV)一起使用,使其既可以直接解释也可以作为三次分析的起点。
(1) 注释说明
gVCF文件的注释包含以下部分:
1) 元信息行以##开头,包含元数据,配置信息,并定义INFO,FILTER和FORMAT字段的值。
2) 标题行以#开头,并命名数据行使用的字段。这些字段是#CHROM,POS,ID,REF,ALT,QUAL,FILTER,INFO,FORMAT,后跟一个或多个样本列。
3) 数据行包含关于基因组中一个或多个位置的信息。
(2) 字段说明
固定字段#CHROM,POS,ID,REF,ALT,QUAL在1000 Genomes Project提供的VCF4.2标准中定义,元信息中描述了字段ID,INFO,FORMAT和样本。
- 1) CHAROM:染色体:来自参考基因组的标识符。
- 2) POS:位置:参考位置,在每个参考序列CHROM中的第一个匹配的起始位置,位置按照递增顺序的数字排序,可以在一个POS中有多个记录。
- 3) ID:可用的唯一标识符。如果没有可用标识符,则使用默认值。
- 4) REF:参考碱基:A,C,G,T,N;可以有多个碱基。POS字段中的值指的是字符串中第一个碱基的位置。
5) ALT:逗号分隔的非等位基因列表,选项是:
- a) 由碱基A,C,G,T,N组成的碱基串。
- b) ID字符串(“”)
- c) 或者是关于break-edns的部分中断替换字符串。
如果没有其他等位基因,则使用缺失值。
- 6) QUAL:用于ALT中断的Phred-scale质量分数。即-10log_10概率。如果未知,则使用默认值。
- 7) FILTER:如果该位置已通过所有过滤器,即在该位置进行call并通过。否则,如果该位置未通过所有过滤器,则以分号分隔的过滤器代码列表表示。
- 8) INFO:其他信息。INFO字段被编码为分号分隔的一些列短键,其中可选值的格式如下:=[, data]。gVCF文件使用以下值:
- a) END:该记录中描述的区域的结束位置。
- b) BLOCKAVG_min30p3a:非变量block块。
- c) SNVSB:SNV位点链偏移
- 9) FORMAT:实例字段的格式。FORMAT指定子字段的数据类型和顺序。gVCF文件使用以下值:
- a) GT:基因型
- b) GQ:基因型质量
- c) GQX:基因型质量假设变异位置,基因型质量假定非变异位置
- d) DP:用于基因分型的过滤call深度。
- e) DPF:在基因分型之前从输入中过滤掉基础调用。
- f) AD:列出顺序中ref和alt等位基因的等位基因深度。对于indels,这个值只包括每个等位基因的读段。
- g) DPI:读取与indel相关的深度,取自indel之前的集合。
10) SAMPLE:样本实例在FORMAT中各个实例字段对应的值。
##fileformat=VCFv4.2 ##ALT=<ID=NON_REF,Description="Represents any possible alternative allele at this location"> ##FILTER=<ID=LowQual,Description="Low quality"> ##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)"> ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##INFO=<ID=MQ,Number=1,Type=Float,Description="RMS Mapping Quality"> ##INFO=<ID=MQRankSum,Number=1,Type=Float,Description="Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities"> ##INFO=<ID=RAW_MQ,Number=1,Type=Float,Description="Raw data for RMS Mapping Quality"> ##INFO=<ID=ReadPosRankSum,Number=1,Type=Float,Description="Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias"> ##contig=<ID=NC_000913.3,length=1769345> ##reference=file:///home/elon/wgs/input/fasta/E.coli_K12_MG1655.fa #CHROM POS ID REF ALT QUAL FILTER INFO FORMAT E.coli_K12_SRR3226034 NC_000913.3 1 . A <NON_REF> . . END=1 GT:DP:GQ:MIN_DP:PL 0/0:8:24:8:0,24,334 NC_000913.3 2 . G <NON_REF> . . END=2 GT:DP:GQ:MIN_DP:PL 0/0:8:0:8:0,0,174 … … NC_000913.3 41 . T C,<NON_REF> 112.77 . BaseQRankSum=1.309;ClippingRankSum=0.000;DP=9;ExcessHet=3.0103;MLEAC=1,0;MLEAF=0.500,0.00;MQRankSum=-0.589;RAW_MQ=32164.00;ReadPosRankSum=-0.100 GT:AD:DP:GQ:PL:SB 0/1:4,5,0:9:57:141,0,57,152,71,223:1,3,5,0 NC_000913.3 46 . T TAA,<NON_REF> 135.73 . BaseQRankSum=0.566;ClippingRankSum=0.000;DP=8;ExcessHet=3.0103;MLEAC=1,0;MLEAF=0.500,0.00;MQRankSum=-0.489;RAW_MQ=28564.00;ReadPosRankSum=1.292 GT:AD:DP:GQ:PL:SB 0/1:3,5,0:8:98:173,0,98,183,113,296:2,1,3,2
上面是对样本SRR3226034的基因测序产生的gVCF文件的简化部分,从注释信息中可以看到文件格式是VCFv4.2 ,基因组用##contig注释信息表明,这里是指ID号为NC_000913.3的染色体,长度为1769345。参考基因组文件通过##reference注释信息标明,这里指的是路径为file:///home/elon/wgs/input/fasta/E.coli_K12_MG1655.fa下的文件。在最后给出的几条样例中,描述了在该物种的NC_000913.3染色体上第1号位点和第2号位点上的两段非变异性调用(分别以A和G开头)。另外最后两个是该物种的NC_000913.3染色体上第41号位点的变异信息和第46号位点的变异信息,这两条信息串的结束位点都是缺失的,因此说明变异信息只包含在该开始位点上。
(4) VCF文件格式
VCF是一种文本文件格式,通常会以“.gz”的压缩方式存储。它包含一个元信息行,一个标题行,然后数据行是包含基因组中一个位点的信息。这个格式也能包含样本组各个位点的基因型信息。
1) 样本组VCF文件
下面给出的VCF文件就是通过九组样本文件分别的变异检测得到的gVCF文件合并得到的,里面包含了该物种的这九组样本所检测到的基因变异位点的信息。VCF文件的格式大致上和gVCF相同,因为它们都是按照The Variant Call Format(VCF)Version 4.2 Specification规范来执行的,唯一不同之处是最后一列样本实例信息列,在单个gVCF文件中都只是包含单个样本文件的信息。而在VCF文件中,由于此处是通过九个gVCF文件合并得到的,因此样本列包含九个样本的所有位点信息。
##fileformat=VCFv4.2
##ALT=<ID=NON_REF,Description="Represents any possible alternative allele at this location">
##FILTER=<ID=LowQual,Description="Low quality">
##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)">
##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##INFO=<ID=MQ,Number=1,Type=Float,Description="RMS Mapping Quality">
##INFO=<ID=MQRankSum,Number=1,Type=Float,Description="Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities">
##INFO=<ID=QD,Number=1,Type=Float,Description="Variant Confidence/Quality by Depth">
##INFO=<ID=RAW_MQ,Number=1,Type=Float,Description="Raw data for RMS Mapping Quality">
##INFO=<ID=ReadPosRankSum,Number=1,Type=Float,Description="Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias">
##INFO=<ID=SOR,Number=1,Type=Float,Description="Symmetric Odds Ratio of 2x2 contingency table to detect strand bias">
##contig=<ID=NC_000913.3,length=1769345>
##reference=file:///home/elon/wgs/input/fasta/E.coli_K12_MG1655.fa
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT E.coli_K12_SRR3226034 E.coli_K12_SRR3226035 E.coli_K12_SRR3226036 E.coli_K12_SRR3226037 E.coli_K12_SRR3226038 E.coli_K12_SRR3226039 E.coli_K12_SRR3226040 E.coli_K12_SRR3226041 E.coli_K12_SRR3226042
NC_000913.3 41 . T C 108 . AC=1;AF=0.071;AN=14;BaseQRankSum=1.31;ClippingRankSum=0.00;DP=234;ExcessHet=3.0103;FS=13.222;MLEAC=1;MLEAF=0.071;MQ=59.78;MQRankSum=-5.890e-01;QD=12.00;ReadPosRankSum=-1.000e-01;SOR=3.590 GT:AD:DP:GQ:PL 0/1:4,5:9:57:141,0,57 0/0:42,0:42:99:0,114,1710 0/0:26,0:26:72:0,72,1080 0/0:33,0:33:99:0,99,1345 ./.:0,0:0:.:0,0,0 ./.:0,0:0:.:0,0,0 0/0:41,0:41:90:0,90,1350 0/0:44,0:44:99:0,105,1575 0/0:39,0:39:99:0,102,1530
NC_000913.3 46 . T TA,TAA 398.53 . AC=2,1;AF=0.143,0.071;AN=14;BaseQRankSum=0.566;ClippingRankSum=0.00;DP=242;ExcessHet=4.1497;FS=0.920;MLEAC=2,1;MLEAF=0.143,0.071;MQ=59.63;MQRankSum=0.00;QD=4.43;ReadPosRankSum=1.29;SOR=0.527 GT:AD:DP:GQ:PL 0/2:3,0,5:8:98:173,183,296,0,113,98 0/1:34,9,0:43:99:166,0,941,268,976,1244 0/0:26,0,0:26:35:0,35,939,35,939,939 0/0:33,0,0:33:99:0,99,1345,99,1345,1345 ./.:0,0,0:0:.:0,0,0,0,0,0 ./.:0,0,0:0:.:0,0,0,0,0,0 0/1:30,5,4:39:41:105,0,798,41,791,1107 0/0:44,0,0:44:99:0,105,1575,105,1575,1575 0/0:39,0,0:39:99:0,102,1530,102,1530,1530
2) 基因变异位点信息
通过一系列的基因测序流程以及和参考基因组的比对,最终得到的VCF文件包含了这九组样本的所有基因变异位点信息。在每个样本得到的gVCF文件,其中包含非变异性调用信息和变异性调用信息,因此整个文件大小在30M左右。而真正的变异位点的片段占很少一部分,因此将九个gVCF文件中变异性调用位点信息合并到一起就小很多,只有3M左右,里面包含的全部是变异调用位点。由于样本的随机性和基因变异的不确定性,可能某位点的变异性只存在与某些样本中,因此在VCF文件中就会看到某些变异片段仅在某些样本列下存在变异信息的情况
2.3 本章小结
这一章主要对构建基于Hadoop的全基因组测序平台所涉及的多方面作了介绍,其中包括Hadoop框架中的HDFS原理、MapReduce框架和YARN框架的介绍;介绍了一个作业在Hadoop集群中运行和调度的全过程;接着对全基因组测序流程的设计作了一个详细的剖析,然后介绍了测序中涉及比较多的两种数据格式FASTA和FASTQ,以及对基因测序得到的中间流程的变异检测结果文件gVCF和最终的变异检测文件VCF作了简要说明。
在接下来的一章中,将会对测序平台中MapReduce程序和测序处理脚本的关键技术做一个更为详细的剖析,例如测序流程是如何与MapReduce框架结合起来的,不同的数据格式是如何进行访问和存储,以及FreeMarker模板引擎的引入和分析流程模板的编写等。
- 蔡斌, 陈湘萍. Hadoop 技术内幕:深入解析Hadoop Common 和HDFS 架构设计与实现原理[M]. 机械工业出版社, 2013. ↩
- 董西成. Hadoop技术内幕:深入解析MapReduce架构设计与实现原理[M]. 机械工业出版社, 2013 ↩
- 董西成. Hadoop技术内幕:深入解析YARN架构设计与实现原理[M]. 机械工业出版社, 2013. ↩
- 陈浩锋. 新一代基因组测序技术[M]. 科学出版社, 2017. ↩
- Richard M, Leggett. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics[R]. US National Library of Medicine, 2013. 4-28. ↩
- FastQC. The FastQC Toolkit[EB/OL]. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/, 2018. ↩
以上是关于2 相关技术及原理的主要内容,如果未能解决你的问题,请参考以下文章