R语言为啥corr.test计算出来的P值全是1
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了R语言为啥corr.test计算出来的P值全是1相关的知识,希望对你有一定的参考价值。
R语言使用corr.test计算相关性矩阵,结果里有一个P值矩阵,但是为什么矩阵的上三角P值全是1而下三角是正常的呢
corr.test默认进行holm法对P值进行校正,上三角是校正后的结果,下三角是原始P值,出现这种情况说明你的显著性校正后没有结果。 参考技术A 以438 226为例binom.test(x =438, n =664, p = 0.5, alternative = "two.sided")
binom.test(x =226, n =664, p = 0.5, alternative = "two.sided")追问
不是太懂,大神可以说的再详细点吗
技术贴 | R语言:组学关联分析和pheatmap可视化
点击蓝字↑↑↑“微生态”,轻松关注不迷路

本文由阿童木根据实践经验而整理,希望对大家有帮助。
原创微文,欢迎转发转载。
举例展示R语言组学关联分析的方法。宏基因组数据以KO-样品丰度表为例。代谢组数据以metabolite-样品丰度表为例。基本方法是用R语言psych包corr.test函数进行两组数据的相关分析,结果经格式化后用pheatmap可视化得热图。
一、模拟输入
1. KO丰度表
代码:
ko_abun = as.data.frame(matrix(abs(round(rnorm(200, 100, 10))), 10, 20))
colnames(ko_abun) = paste("KO", 1:20, sep="_")
rownames(ko_abun) = paste("sample", 1:10, sep="_")
ko_abun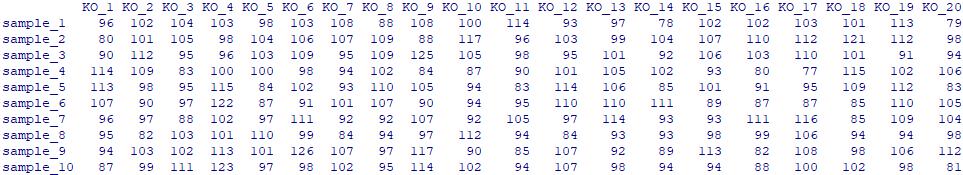
图 1
2. metabolite丰度表
代码:
metabo_abun = as.data.frame(matrix(abs(round(rnorm(200, 200, 10))), 10, 20))
colnames(metabo_abun) = paste("met", 1:20, sep="_")
rownames(metabo_abun) = paste("sample", 1:10, sep="_")
metabo_abun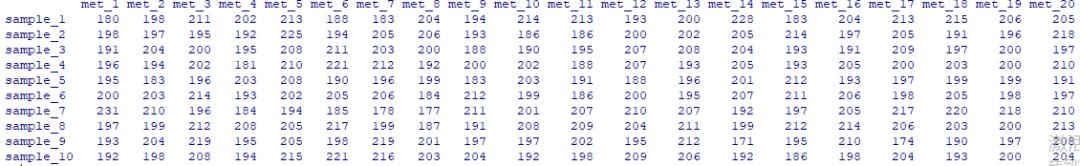
图 2
二、相关分析函数
思路
参数一:other -> KO或其他组学丰度表
参数二:metabo -> 代谢物丰度表
参数三:route -> 输出目录【提前创建】
corr.test进行两组数据相关分析
用stringr split将ko-metabolite结果列拆成两列
结果保留r_value p_value
显著相关标记“*”
library(psych)
library(stringr)
correlate = function(other, metabo, route)
{
# 读取方式:check.name=F, row.names=1, header=T
# 计算相关性:
result=data.frame(print(corr.test(other, metabo, use="pairwise", method="spearman", adjust="fdr", alpha=.05, ci=TRUE, minlength=50), short=FALSE, digits=5))
# 整理结果
pair=rownames(result) # 行名
result2=data.frame(pair, result[, c(2, 4)]) # 提取信息
# P值排序
result3=data.frame(result2[order(result2[,"raw.p"], decreasing=F),])
# 格式化结果【将细菌代谢物拆成两列】
result4=data.frame(str_split_fixed(result3$pair, "-", 2), result3[, c(2, 3)])
colnames(result4)=c("feature_1", "feature_2", "r_value", "p_value")
# 保存提取的结果
write.table(result4, file=paste(route, "Correlation_result.txt", sep="/"), sep=" ", row.names=F, quote=F)
}
三、相关性分析
代码
dir.create("Result") # 创建结果目录
correlate(ko_abun, metabo_abun, "Result")
结果Correlation_result.txt如下,行数为20X20
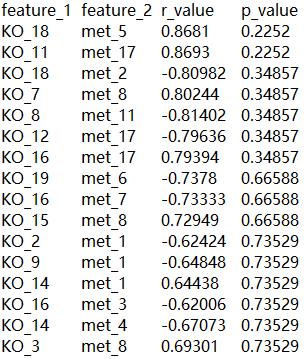
图3
四、pheatmap可视化函数
思路:
参数一:infile ->Correlation_result.txt相关性分析结果
参数二:route -> Route输出路径
用reshape2 dcast函数把feature_1 feature_2 p_value做成matrix,作为pheatmap输入文件
用reshape2 dcast函数把feature_1 feature_2 r_value做成matrix,作为pheatmap display
代码:
library(reshape2)
library(pheatmap)
correlate_pheatmap = function(infile, route)
{
data=read.table(paste(route, infile, sep="/"), sep=" ", header=T)
data_r=dcast(data, feature_1 ~ feature_2, value.var="r_value")
data_p=dcast(data, feature_1 ~ feature_2, value.var="p_value")
rownames(data_r)=data_r[,1]
data_r=data_r[,-1]
rownames(data_p)=data_p[,1]
data_p=data_p[,-1]
# 用"*"代替<=0.05的p值,用""代替>0.05的相对丰度
data_mark=data_p
for(i in 1:length(data_p[,1])){
for(j in 1:length(data_p[1,])){
data_mark[i,j]=ifelse(data_p[i,j] <= 0.05, "*", "")
}
}
pheatmap(data_r, display_numbers=data_mark, cellwidth=20, cellheight=20, fontsize_number=18, filename=paste(route, "Correlation_result.pdf", sep="/"))
pheatmap(data_r, display_numbers=data_mark, cellwidth=20, cellheight=20, fontsize_number=18, filename=paste(route, "Correlation_result.png", sep="/"))
}
五、pheatmap绘制热图
代码:
correlate_pheatmap("Correlation_result.txt", "Result")结果目录,新增结果图(一个png一个pdf):
图4
打开pdf,如下:
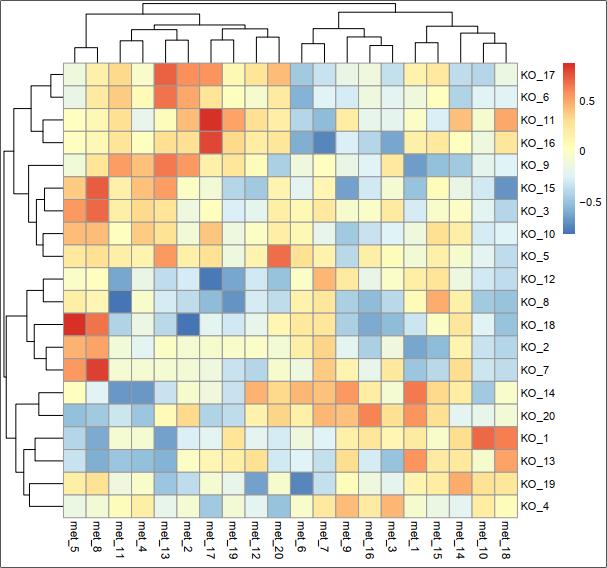
图5
随机模拟的数据,没有显著的不奇怪。如果是做项目一般两组数据的相关分析都可以得到一些显著相关的结果,如下:
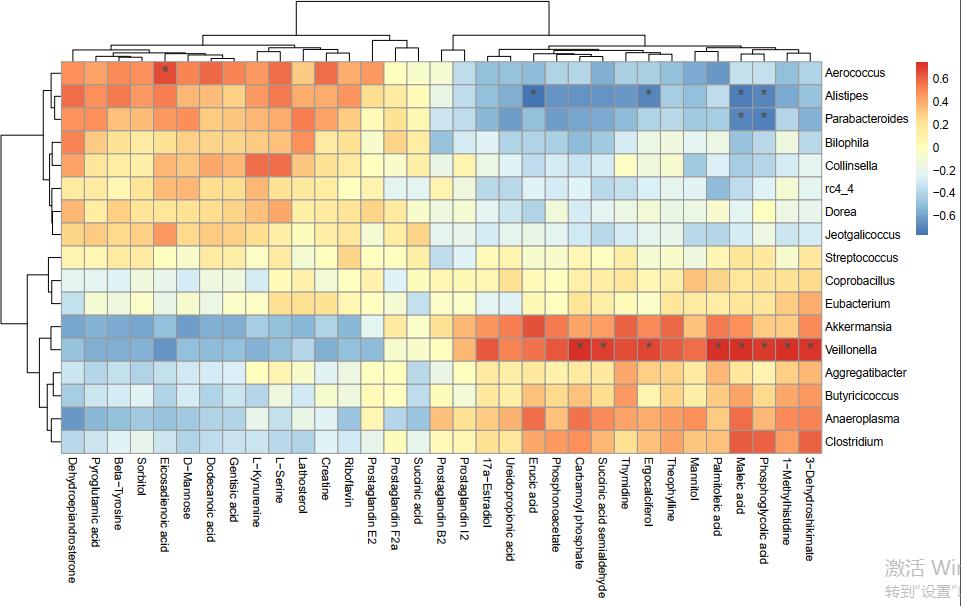
图6
 你可能还喜欢
你可能还喜欢
1
4
5
微生态科研学术群期待与您交流更多微生态科研问题
(联系微生态老师即可申请入群)
了解更多菌群知识,请关注“微生态”。
以上是关于R语言为啥corr.test计算出来的P值全是1的主要内容,如果未能解决你的问题,请参考以下文章