WGCNA(转载)
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了WGCNA(转载)相关的知识,希望对你有一定的参考价值。
参考技术A WGCNA原理及应用WGCNA介绍:
WGCNA(weighted gene co-expression network analysis,权重基因共表达网络分析)是一种分析多个样本基因表达模式的分析方法,可将表达模式相似的基因进行聚类,并分析模块与特定性状或表型之间的关联关系,因此在疾病以及其他性状与基因关联分析等方面的研究中被广泛应用。
WGCNA算法是构建基因共表达网络的常用算法(详解: http://www.jianshu.com/p/94b11358b3f3 )。WGCNA算法首先假定基因网络服从无尺度分布,并定义基因共表达相关矩阵、基因网络形成的邻接函数,然后计算不同节点的相异系数,并据此构建分层聚类树(hierarchical clustering tree),该聚类树的不同分支代表不同的基因模块(module),模块内基因共表达程度高,而分属不同模块的基因共表达程度低。最后,探索模块与特定表型或疾病的关联关系,最终达到鉴定疾病治疗的靶点基因、基因网络的目的。在该方法中module被定义为一组具有类似表达谱的基因,如果某些基因在一个生理过程或不同组织中总是具有相类似的表达变化,那么我们有理由认为这些基因在功能上是相关的,可以把他们定义为一个模块(module)。这似乎有点类似于进行聚类分析所得到结果,但不同的是,WGCNA的聚类准则具有生物学意义,而非常规的聚类方法(如利用数据间的几何距离),因此该方法所得出的结果具有更高的可信度。当基因module被定义出来后,我们可以利用这些结果做很多进一步的工作,如关联性状,代谢通路建模,建立基因互作网络等。
WGCNA的用处:
这类处于调控网络中心的基因称为核心基因(hub gene),这类基因通常是转录因子等关键的调控因子,是值得我们优先深入分析和挖掘的对象。
在网络中,被调控线连接的基因,其表达模式是相似的。那么它们潜在有相似的功能。所以,在这个网络中,如果线条一端的基因功能是已知的,那么就可以预测线条另一端的功能未知的基因也有相似的功能。
下面的问答来自基迪奥,也能加深对WGCNA的理解
问1、调控网络和共表达网络有什么区别?
答:调控网络是个更广泛的概念,而共表达网络是调控网络的一种。
理论上我们可以利用各类信息构建调控网络(表达相关性,序列靶向关系、蛋白互作关系),另外调控网络构建的信息既可以来源真实的实验验证的关系,也可以来源生物信息的预测。而共表达网络特指利用基因间的表达相关性预测基因间调控关系的方法,而WGCNA又是共表达网络分析中最有效的方法之一。
问2、WGCNA分析适合的生物物种范围有规定么?
答:没有限制。对于任何物种中心法则都是存在的,调控关系对于任何物种都是存在的,所以WGCNA没有物种限定。
问3、同一物种,不同来源的转录组数据(比如不同文章/资料来源的),可以放在一起做WGCNA分析吗?
答:只要样本间有相似的生物学意义,是可以合并在一起做分析的。但要注意,不同批次之间的样本是有批次效应的,所以可能会带来一些误差,但是是可以放在一起分析的。
问4、相同材料不同处理之间,可以放在一起做WGCNA分析吗?比如重金属和盐碱处理。
答:可以的。这也正式WGCNA强大的地方,其可以将不同处理的样本,合并在一起做分析。其他方法则不一定有这么强大的能力,比如做基因表达趋势分析时,如果样本涉及到多个处理不同时期的时候,就不好合并分析(或合并后难以解读)。但WGCNA的方法关注的是调控关系,所以不管是多少个处理组,都可以很好的整合在一起做分析。
问5、不同批次的数据能放一起做WGCNA吗?
答:可以的。虽然有批次的干扰,但是干扰对WGCNA网络没有太大影响。因为WGCNA不是做差异分析,而是基因的共表达。因为批次效应理论上不影响相关性。
问6、不同类型的材料,比如亲本和F1,适合放一起进行WGCNA么?
答:如果是一个作图群体,当然亲本与F1是可以放在一起分析的,因为你只关心基因的表达模式,所以把亲本加进来是没有问题的。
问7、没有生物学重复,共3组,每组5个时间点能够做吗?
答:理论上有15个样本,是可以做WGCNA分析的。并且,分析出来的结果对你的研究应该是非常有用的。至少他会比趋势分析更有意义,更加准确。
问8、一般说WGCNA的样品不少于15个,15个样品考虑重复吗?不同倍性的材料呢?
答:15个样本这个是包含了生物学重复,比如5个时间点3个重复;在RNA-seq里面建议不要用不同倍性材料加进来。除非是有参考的多倍体,如果是无参的多倍体,不同倍性之间差异太大,会让调控网络不准确。所以用单一倍性的材料做调控网络会更加准确。
问9、可以将RNA-seq数据与蛋白组数据,甲基化数据放一起做WGCNA分析?
答:不能与蛋白数据一起分析。因为WGCNA是基于相关系数的算法。所以最好一起分析的数据变异度是类似的,RNAseq变异非常大,而蛋白的数据变异很小,两者的变化不在一个数量级上面。所以两种数据放在一起分析不合理。
但RNA数据可以尝试跟甲基化数据一起分析。当然我们也建议RNA数据与代谢组数据一起分析,因为代谢组的数据变异也非常大。
问10、表达量和表达的基因数目差异太大的样品可以一起分析吗?比如样品A有2k个gene表达 而样品B有2w个gene表达了 AB可以一起分析吗?
答:做WGCNA分析的时候,不能脱离生物学意义,既然要分析调控网络,那么应该分析有相似生物学意义的一组基因,比如说拿相似组织来一起做分析,比如不应该拿大脑的样本与脚趾的样本合并在一起做分析,因为很显然,这两个组织没有关联。如果两个样本之间是有相关联的生物学意义,哪怕表达的基因数不一样,或表达模式差异很大,那依然可以放在一起分析;但如果样本之间完全没有生物学意义,那么分析就没有意义。
问11、实验设计是case3个时间点(各点都有三个重复),control同样的3个时间点(每点三个重复),WGCNA怎么做?3个时间点和case-control两个因素能同时考虑进来分析吗?
答:可以的。做WGCNA是更加合理的,因为有两个梯度的样本,如果只是做差异分析的话,逻辑可能非常复杂,做WGCNA分析是对样本特性更好的解析,可以直观看到基因在六个处理组里面是怎样表达的。
问12、可以拿混合样本分析吗?比如一个病原细菌跟人类细胞的基因,能说明细菌跟人类细胞基因有调控关系吗?
答:可以。前提是病原菌有足够的数据并定量准确,并且这个分析是非常有意义的,最后可以说明这些病原菌可以调控哪些宿主基因。
问13、但是病原宿主混合分析的话,宿主蛋白不能分泌到宿主体内岂不是WGCNA生物学上也没有意义吗?
答:依然有意义。即使病原的基因没有分泌到宿主里面,但是病原的蛋白是会影响宿主基因的调控的,比如某个细菌感染某个植物,虽然细菌的蛋白不能直接分泌到植物体内,但会影响植物蛋白的分泌。混在一起分析依然是有意义,可以看到植物里面到底哪个基因对细菌蛋白产生应答作用。
问14、芯片数据两分类,每组20个样本,能否每组单独做WGCNA?
答:可以。WGCNA还有一种重要功能是做两个网络的比较,比如病人20个样本做一个调控网络,健康人做一个调控网络,然后两个网络做比较。
问15、WGCNA可以用来分析lncRNA对下游基因的调控分析吗?
答:可以。WGCNA网络有利于预测lncRNA的潜在功能。
问16、构建网络是用所有表达基因还是差异基因?
答:这个是具体问题具体分析。如果使用所有的基因分析,会导致运算量非常大。而也不是所有的基因在这个实验中都有生物学意义,所以我们会提前做一些过滤。
但用于分析的基因不一定是差异表达基因,有时可以用差异表达基因做一个并集,或通过计算变异系数将变异系数低的基因以及低表达的基因去除。但注意,如果你有关心的特定目标基因的话,应该尽量给予保留。
问17、关注某一个pathway上的基因以及调控因子之间的相关性,构建WGCNA网络的时候属于这个pathway的基因数量太少会不会影响结果呢?
答:这不是问题。在一个调控网络里面,样本的某个pathway上,并不是所有基因参与调控(或存在差异性),所以在做WGCNA分析的时候,会做一些过滤,将有变化的基因挑出来再做分析。即分析的是某个pathway上有变化的基因,不需要分析pathway上所有的基因,只需要分析那些变化的基因就够了。
问18、前期筛选的时候,要选出在所有样本中变异系数比较大的基因呢?还是直接用差异表达的基因取并集?用基因还是转录本,哪个好呢?
答:两则都可以,我推荐使用变异系数,选择那些变异较大的基因,来做下面的分析。然后建议用基因不要用转录本,因为转录本的定量是不准确的。
问19、变异系数一般取多大?
答:具体问题具体分析。例如,没有特定目标的时候,可以先计算变异系数,将变异系数的百分之前50来做分析,把变异系数偏低的后面一半过滤掉。
问20、输入数据用FPKM合适吗?
答:可以。
问21、RNA seq数据是RSEM值怎么办?
答:RSEM值原始输出结果为reads数,如果是RSEM值建议做一个RPKM校正再做分析。
问22、除了RPKM值以外,做WGANA是否还需要其他数据?TCGA数据可否来做WGCNA分析?
答:在做WGCNA分析必须要用表达量数据,但TCGA的数据某些层级没有表达量数据,没有表达量数据自然就无法做WGCNA分析。
问23、请问输入的基因样本的矩阵的时候,要不要对数据标准化?
答:做WGCNA分析的时候,不需要对数据进行标准化,输入RPKM值就足以做这个分析。虽然一些文章会做log2处理,但我认为取了LOG2后,会让一些表达关系没有那么丰富。
问24、每个样本有3个生物学重复,不需要对三个重复的表达量求平均值代表该样本吗?
答:注意,做WGCNA的时候每个样本是独立的,三个生物学重复样本是全部导入做分析,不是取均值再做分析,每个样本都是独立的。
问25、如果3个生物学重复,做WGCNA的时候是取三个值,还是用cuffdiff处理后取一个值?
答:如果是生物学重复样本进行调控网络分析,每个样本独立使用,而不是取均值。
问26、请问将样本信息同模块特征值进行相关性分析的时候,样本信息是怎么处理的呢?比如不同取样点、不同性别什么的,这不是数量性状信息的,这种情况应该怎么处理呢?
答:样本的任何信息都可以做模块相关性分析。比如相关时间点,可以按照先后量化为12134567。又如不同性别,男与女,可以定义为1,-1。任何性状量化为数字后,都可以进行相关性分析。
问27、怎么将模块与性状对应起来呢有些性状不好量化,如果直接将模块与分组对应,如何实现, 不需要量化指标么?
答:首先需要将性状量化,如果无法将性状量化,那么就无法分析。至于分组信息,也可以量化为类似00001111000(1代表一种组别,2代表另一组组别),实现分组信息的数字化。
问28、基因数量为3w左右时,modules数量为多少结果较为理想?怎么评价聚类效果的好坏?
答:modules数量没有标准,modules数量无法评估模块分的好坏,分组是否合理应该看树的树形图,比如树的分支很清晰就说明模块式清晰的。modules数量数由生物性状决定的。比如样本表达信息很丰富的时候,modules数量会很多;如果样本的基因表达相对单一,modules数量就会比较少。
问29、我运行例子的时候,得出来基因之间的direction全是undirected,这和前面的几种关系有什么区别?
答:WGCNA是一个undirected的方法,它的网络是无方向的,有相关关系但是无方向。
问30、如果做有向网络的构建,您推荐那些方法?
答:很多方法,例如贝叶斯的方法。
问31、非模式物种可以得出基因之间的相互关系类型么?得出的结果也是undirected么?
答:WGCNA是基于表达两处理的,所以即使是非模式生物,当然也可以他们之间关系,并且关系也是一个无向网络。
问32、选择几个表型数据进行结合分析比较好
答:越多越好,看实验设计。
问33、感染小鼠,5个时间点,3个重复,找不到合适的表型怎么办?
答:如果找不到合适表型,可以找某个时间点应答的基因,本身基因的表达趋势已经有某种生物学意义的。没有找到合适表型,也可以看变化趋势。不一定要做表型的相关分析,其他分析也是很有趣的。例如,可以对模块功能的富集分析,其实都是可以帮助你找到特定模块的。所以不用纠结于做某个表型的关联分析。
问34、weight就是tom值吗?
答:是的。
问35、剪模块是怎么做的?是根据TOM划分吗?需要自己设定,还是R自动的?
答:剪模块是R中自动完成的,不需要划分,但合并的时候你可以设定一个指标,比如差异度是0.25。
问36、看WGCNA说明是用相异矩阵D(D=1-TOM)去做聚类,然后动态剪切?
答:用TOM值来构建矩阵,TOM值就是两个样本的相似度,1-TOM值就是两个样本的差异度,相似度与差异度可以理解为一个东西,并不矛盾。
问37、模块特征值和样本性状相关分析的具体方法是?
答:R包用的是计算相关系数的方法。
问38、WGCNA里面一般会提到hubgene,如何确定hubgene?
答:在WGCNA分析里面,每个基因都会计算连通性,连通性高的就是hubgene。
问39、在R中安装“”WGCNA“”说不适合R3.3.1,那适合哪个版本?
答:WGCNA应该是所有版本都适合,如果版本没有可以考虑降低R软件的版本,这个对分析没有影响。因为不同R版本是一样的。
问40、用STEM分析的时候拟合多少个模型合适?
答:建议不要超过20个。模块太多不好分析。
参考网站:
http://tiramisutes.github.io/2016/09/14/WGCNA.html
http://www.jianshu.com/p/94b11358b3f3
http://www.omicshare.com/class/home/index/classdetail?id=20
R 语言 4.2.2安装 WGCNA
文章目录
1 WGCNA库介绍
-
WGCNA是用于加权相关网络分析的R包,相关网络越来越多地用于生物信息学应用 -
加权基因共表达网络分析是一种系统生物学方法,用于描述微阵列样本中基因之间的相关性模式 -
加权相关网络分析(WGCNA)可用于发现高度相关基因的簇(模块),使用模块特征基因或模块内中枢基因总结此类簇,可以将模块彼此关联并与外部样本特征关联(使用特征基因网络方法),以及计算模块成员度量 -
相关网络促进了基于网络的基因筛选方法,可用于识别候选生物标志物或治疗靶点 -
这些方法已成功应用于各种生物学背景,例如癌症、小鼠遗传学、酵母遗传学和脑成像数据分析
-
WGCNA是R函数的综合集合,用于执行加权相关网络分析的各个方面。该软件包包括网络构建、模块检测、基因选择、拓扑特性计算、数据模拟、可视化以及与外部软件接口的功能
2 安装
踩坑
直接安装WGCNA,这样的方式会导致一些依赖包安装失败,导致WGCNA运行不起来;
install.packages('WGCNA')
报错信息:
Warning in install.packages :
没有'‘impute’, ‘preprocessCore’, ‘AnnotationDbi’'这种相依关系
trying URL 'https://cran.rstudio.com/bin/windows/contrib/4.2/WGCNA_1.71.zip'
Content type 'application/zip' length 3254830 bytes (3.1 MB)
downloaded 3.1 MB
---------------------------------------------------
> library(WGCNA)
载入需要的程辑包:dynamicTreeCut
载入需要的程辑包:fastcluster
载入程辑包:‘fastcluster’
The following object is masked from ‘package:stats’:
hclust
Error: package or namespace load failed for ‘WGCNA’ in loadNamespace(i, c(lib.loc, .libPaths()), versionCheck = vI[[i]]):
不存在叫‘AnnotationDbi’这个名字的程辑包
>
> library(GO.db)
Error: 找不到‘GO.db’所需要的程辑包‘AnnotationDbi’
> library(WGCNA)
Error: package or namespace load failed for ‘WGCNA’ in loadNamespace(i, c(lib.loc, .libPaths()), versionCheck = vI[[i]]):
不存在叫‘AnnotationDbi’这个名字的程辑包
对相应的依赖单独安装之后,仍然出现相关的问题,而且出现版本不匹配的问题,不能解决问题
还得是官方文档
link: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA
由于作者基本不会R语言,所以不得不查找官方的文档,直接搜索WGCNA会有很多资料,找到官方文档
- 自动安装方法(本人尝试后失败)
install.packages("BiocManager")
BiocManager::install("WGCNA")
如果
BiocManager已经安装的情况下,第一个命令将被跳过
- 手动安装相关的包
install.packages(c("matrixStats", "Hmisc", "splines", "foreach", "doParallel", "fastcluster", "dynamicTreeCut", "survival", "BiocManager"))
BiocManager::install(c("GO.db", "preprocessCore", "impute"));
如果版本较老没有BiocManager,使用:
install.packages(c("matrixStats", "Hmisc", "splines", "foreach", "doParallel", "fastcluster", "dynamicTreeCut", "survival"))
source("http://bioconductor.org/biocLite.R")
biocLite(c("GO.db", "preprocessCore", "impute"))
这样安装我出现的问题
> library(WGCNA)
载入需要的程辑包:dynamicTreeCut
载入需要的程辑包:fastcluster
载入程辑包:‘fastcluster’
The following object is masked from ‘package:stats’:
hclust
Error: package or namespace load failed for ‘WGCNA’ in loadNamespace(j <- i[[1L]], c(lib.loc, .libPaths()), versionCheck = vI[[j]]):
不存在叫‘impute’这个名字的程辑包
问题原因:自从R版本 2.14.0起,CRAN已经将impute撤回,需要通过Biocondutor获取,在R shell中运行
source("http://bioconductor.org/biocLite.R")
biocLite("impute")
# 或者
BiocManager::install(c("GO.db", "preprocessCore", "impute"))
我在 R-4.2.2 中安装暂时没有出现其他ERROR
完成后信息:
> source("http://bioconductor.org/biocLite.R")
Error: With R version 3.5 or greater, install Bioconductor packages using BiocManager; see https://bioconductor.org/install
> BiocManager::install(c("GO.db", "preprocessCore", "impute"))
'getOption("repos")' replaces Bioconductor standard repositories, see '?repositories'
for details
replacement repositories:
CRAN: https://cran.rstudio.com/
Bioconductor version 3.16 (BiocManager 1.30.19), R 4.2.2 (2022-10-31 ucrt)
Installing package(s) 'preprocessCore', 'impute'
trying URL 'https://bioconductor.org/packages/3.16/bioc/bin/windows/contrib/4.2/preprocessCore_1.60.1.zip'
Content type 'application/zip' length 169168 bytes (165 KB)
downloaded 165 KB
trying URL 'https://bioconductor.org/packages/3.16/bioc/bin/windows/contrib/4.2/impute_1.72.2.zip'
Content type 'application/zip' length 667393 bytes (651 KB)
downloaded 651 KB
程序包‘preprocessCore’打开成功,MD5和检查也通过
程序包‘impute’打开成功,MD5和检查也通过
参考
WGCNA官网: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/
WGCNA包安装FAQ:https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/faq.html
Appendix
A. 安装R
下载 R-windows 以及 RStudio :
R语言Windows安装: https://blog.csdn.net/weixin_44524441/article/details/114130789
安装RStudio: https://blog.csdn.net/qq_34848334/article/details/119762092
B. 配置环境
修改R语言环境:
RStudio -> Tools -> Global Options 中换掉相关的环境,并重启RStudio
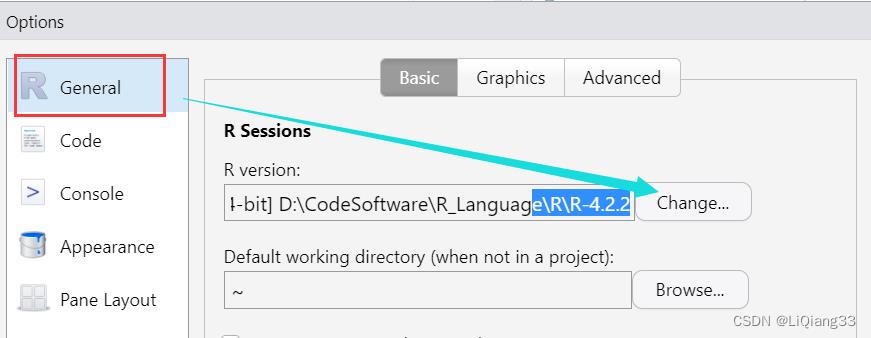
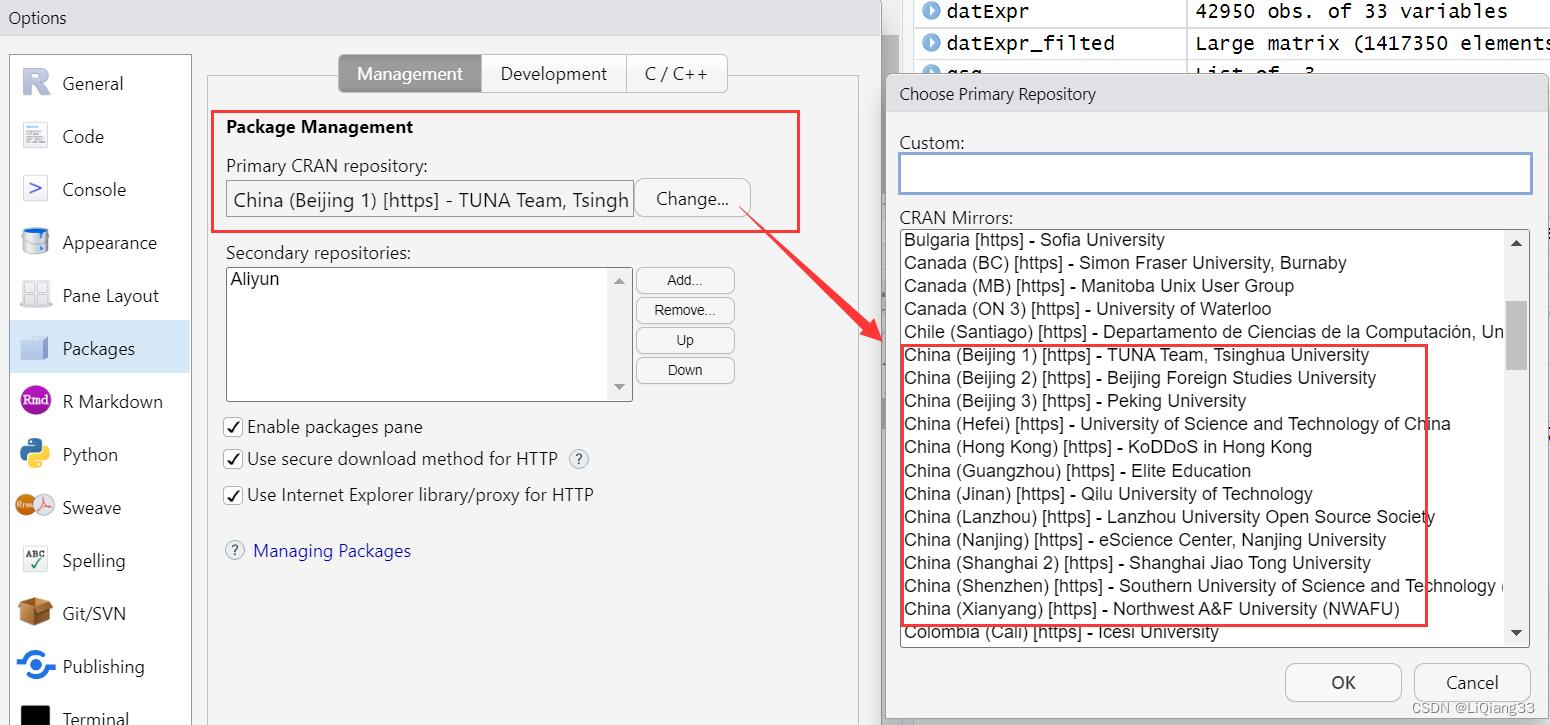
C. 修改镜像
RStudio -> Tools -> Global Options 中 Packages 选择中国镜像
以上是关于WGCNA(转载)的主要内容,如果未能解决你的问题,请参考以下文章