系统生物学大肠杆菌蛋白互作网络分析(系统与合成生物学第三次作业)
Posted Dream of Grass
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了系统生物学大肠杆菌蛋白互作网络分析(系统与合成生物学第三次作业)相关的知识,希望对你有一定的参考价值。
摘要
下载大肠杆菌蛋白互作网络(Ecoli PPI network)数据,使用Python对大肠杆菌蛋白互作网络进行筛选,并使用Cytoscape进行圆形布局可视化。此外,还绘制度分布函数并用幂函数进行拟合。
大肠杆菌蛋白互作网络
数据下载
我们从string(https://cn.string-db.org/)中下载ppi数据。
- 大肠杆菌蛋白互作网络下载地址:https://stringdb-static.org/download/protein.links.v11.5/511145.protein.links.v11.5.txt.gz
- 蛋白质信息下载地址:https://stringdb-static.org/download/protein.info.v11.5/511145.protein.info.v11.5.txt.gz
数据处理
将蛋白质的原始id转换为蛋白质名,并筛选结合分数大于800的蛋白质对。
import pandas as pd
import networkx as nx
# 处理Ecoli的ppi数据
filepath = "D:/000大三下/系统与合成生物学/"
link_filename = '511145.protein.links.v11.5.txt'
info_filename = '511145.protein.info.v11.5.txt'
ppi_link = pd.read_csv(filepath + link_filename, sep=' ', header=0)
ppi_info = pd.read_csv(filepath + info_filename, sep='\\t', header=0)
# 筛选结合分数大于800的protein pair
ppi_link = ppi_link[ppi_link['combined_score'] > 800]
# 把stiring_protein_id替换成preferred_name
ppi = pd.merge(ppi_link, ppi_info[['#string_protein_id', 'preferred_name']], left_on='protein1',
right_on='#string_protein_id')
ppi = pd.merge(ppi, ppi_info[['#string_protein_id', 'preferred_name']], left_on='protein2',
right_on='#string_protein_id')
ppi = ppi[['preferred_name_x', 'preferred_name_y', 'combined_score']]
ppi.columns = ['protein1', 'protein2', 'combined_score']
# 保存
ppi.to_csv(filepath + 'Ecoli_ppi.csv', index=False, header=1)
处理后的数据如下:
| protein1 | protein2 | combined_score |
|---|---|---|
| thrA | dapA | 945 |
| dapB | dapA | 999 |
| dapD | dapA | 966 |
| dapE | dapA | 948 |
| bamC | dapA | 945 |
| lysA | dapA | 984 |
| asd | dapA | 994 |
| dapF | dapA | 968 |
| metL | dapA | 938 |
| lysC | dapA | 858 |
圆形布局可视化蛋白互作网络

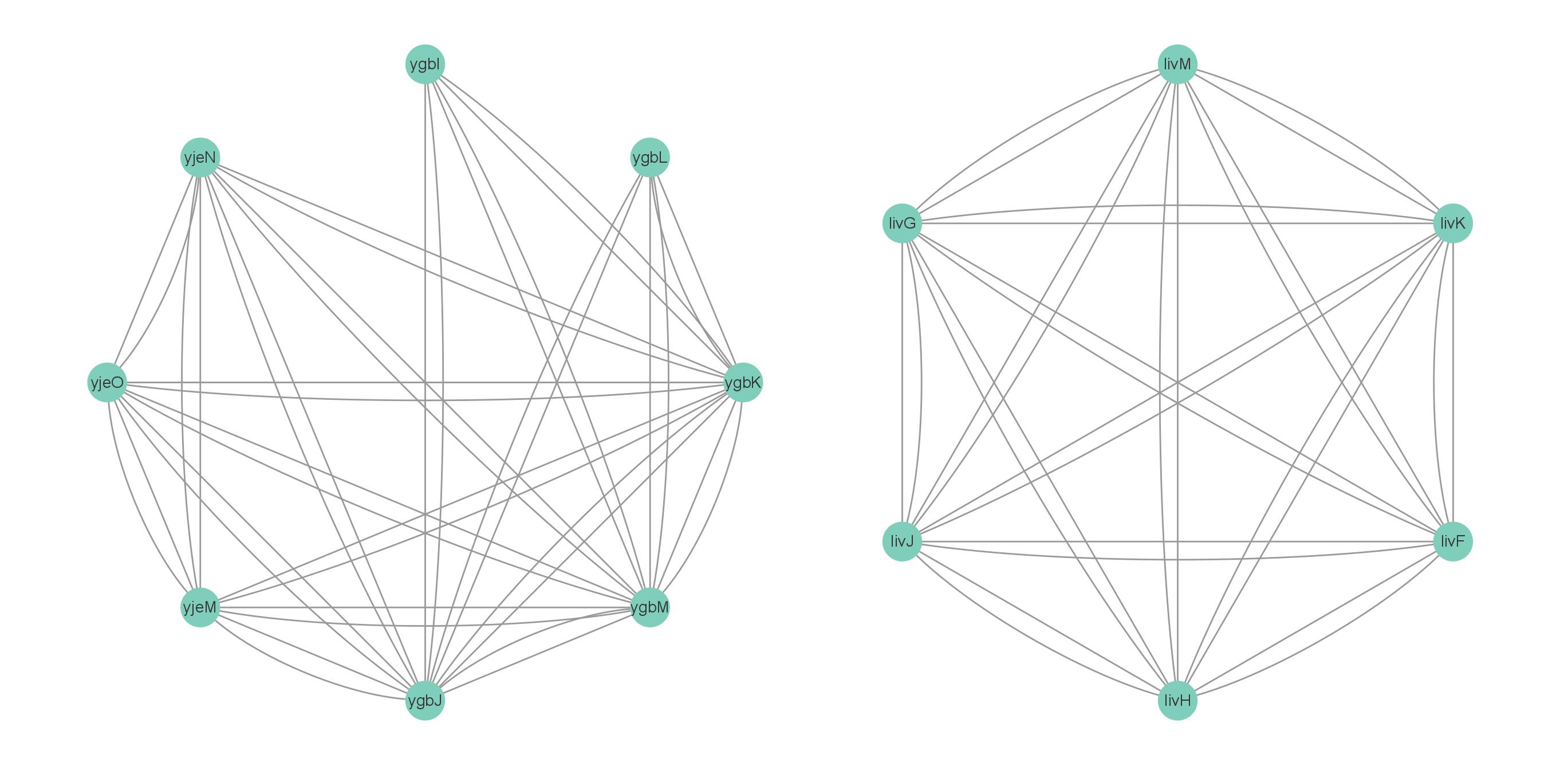
计算中心性指标
使用networkx,计算Degree Centrality、Closeness Centrality、Betweenness Centrality和Eigenvector Centrality。计算结束后对其求和并降序排列。
net = ppi[['protein1', 'protein2']]
net.drop_duplicates()
G = nx.from_pandas_edgelist(net, "protein1", "protein2")
# 计算中心性
degree_centrality = nx.degree_centrality(G)
closeness_centrality = nx.closeness_centrality(G)
betweenness_centrality = nx.betweenness_centrality(G)
eigenvector_centrality = nx.eigenvector_centrality(G)
# 将指标转换为DataFrame格式
centrality_df = pd.DataFrame('Degree Centrality': degree_centrality,
'Closeness Centrality': closeness_centrality,
'Betweenness Centrality': betweenness_centrality,
'Eigenvector Centrality': eigenvector_centrality)
# 求和并排序
centrality_df['Sum'] = centrality_df.sum(axis=1)
centrality_df = centrality_df.sort_values(by='Sum', ascending=False)
# 保存
centrality_df.to_csv(filepath + 'Ecoli_ppi_centrality.csv')
| Protein | Degree Centrality | Closeness Centrality | Betweenness Centrality | Eigenvector Centrality | Sum |
|---|---|---|---|---|---|
| rpoB | 0.032583 | 0.274943 | 0.018549 | 0.105043 | 0.431118 |
| rpsL | 0.029222 | 0.260888 | 0.011465 | 0.115507 | 0.417082 |
| rplB | 0.029222 | 0.261492 | 0.00294 | 0.121612 | 0.415266 |
| rpmA | 0.027411 | 0.263495 | 0.006903 | 0.117347 | 0.415155 |
| rplM | 0.029222 | 0.25926 | 0.002129 | 0.121699 | 0.41231 |
| rplE | 0.028963 | 0.257817 | 0.002343 | 0.122201 | 0.411324 |
| rpsB | 0.027929 | 0.260983 | 0.002664 | 0.117817 | 0.409392 |
| rplD | 0.028187 | 0.256263 | 0.001058 | 0.122461 | 0.407969 |
| rplV | 0.027929 | 0.255628 | 0.001112 | 0.121178 | 0.405846 |
| rplC | 0.02767 | 0.255719 | 0.00102 | 0.12137 | 0.405779 |
绘制度分布函数并用幂函数拟合
使用python进行绘制并拟合。幂指数为-0.599。
# 计算度分布函数
# 即每个度数对应的节点数除以总节点数。
degree_sequence = sorted([d for n, d in G.degree()], reverse=True)
degree_count = np.zeros(max(degree_sequence)+1)
for degree in degree_sequence:
degree_count[degree] += 1
degree_distribution = degree_count/sum(degree_count)
# 绘制度分布函数的双对数轴图
x = np.arange(1, len(degree_distribution)+1)
y = degree_distribution
# plt.loglog(x, y, color = 'chocolate', marker = '.')
plt.plot(x, y, color = 'chocolate', marker = '.')
# 使用幂函数拟合,计算幂指数的值
def power_law(x, a, b):
return a * x ** (b)
popt, pcov = curve_fit(power_law, x, y)
a, b = popt
# plt.loglog(x, power_law(x, a, b), 'c--', label='fit: y=%5.3f*x^(%5.3f)' % (a, b))
plt.plot(x, power_law(x, a, b), 'c--', label='fit: y=%5.3f*x^(%5.3f)' % (a, b))
plt.xlabel('Degree)')
plt.ylabel('Frequency)')
# plt.xlabel('log(Degree)')
# plt.ylabel('log(Frequency)')
plt.legend()
plt.savefig(filepath+'Degree_distribution.png')
plt.show()
print('幂指数的值为:', b)
 | 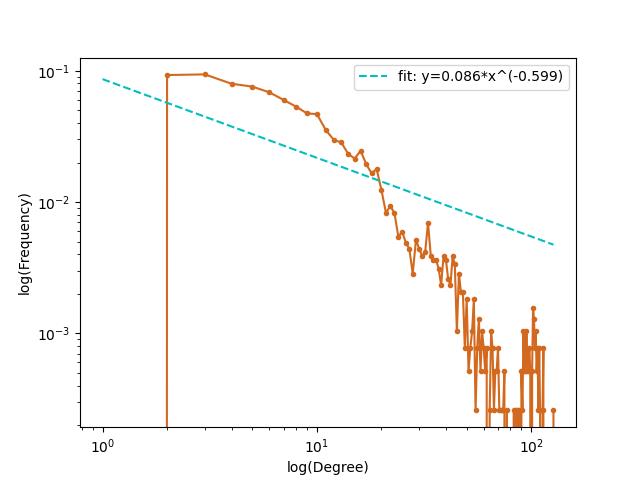 |
| 图3 横纵坐标对数转换前 | 图4 横纵坐标对数转换后 |
完整代码
# _*_ coding: utf-8 _*_
# @Time : 2023-03-28 23:03
# @Author : YingHao Zhang(池塘春草梦)
# @Version:v0.1
# @File : network.py
# @desc : 系统与合成生物学作业。读取Ecoli的ppi数据,将蛋白质的原始id转换为蛋白质名,并计算四种中心性指标。输入:ppi的link和info文件。输出:①处理后的ppi网络(包括蛋白质对和结合分数);②蛋白质的四种中心性指标和四种中心性指标的和。绘制度分布曲线,并用幂函数拟合。
import pandas as pd
import networkx as nx
import time
start_time = time.time()
# 处理Ecoli的ppi数据
filepath = "D:/000大三下/系统与合成生物学/"
link_filename = '511145.protein.links.v11.5.txt'
info_filename = '511145.protein.info.v11.5.txt'
ppi_link = pd.read_csv(filepath + link_filename, sep=' ', header=0)
ppi_info = pd.read_csv(filepath + info_filename, sep='\\t', header=0)
# 筛选结合分数大于800的protein pair
ppi_link = ppi_link[ppi_link['combined_score'] > 800]
# 把stiring_protein_id替换成preferred_name
ppi = pd.merge(ppi_link, ppi_info[['#string_protein_id', 'preferred_name']], left_on='protein1',
right_on='#string_protein_id')
ppi = pd.merge(ppi, ppi_info[['#string_protein_id', 'preferred_name']], left_on='protein2',
right_on='#string_protein_id')
ppi = ppi[['preferred_name_x', 'preferred_name_y', 'combined_score']]
ppi.columns = ['protein1', 'protein2', 'combined_score']
# 保存
ppi.to_csv(filepath + 'Ecoli_ppi.csv', index=False, header=1)
net = ppi[['protein1', 'protein2']]
net.drop_duplicates()
G = nx.from_pandas_edgelist(net, "protein1", "protein2")
# 计算中心性
degree_centrality = nx.degree_centrality(G)
closeness_centrality = nx.closeness_centrality(G)
betweenness_centrality = nx.betweenness_centrality(G)
eigenvector_centrality = nx.eigenvector_centrality(G)
# 将指标转换为DataFrame格式
centrality_df = pd.DataFrame('Degree Centrality': degree_centrality,
'Closeness Centrality': closeness_centrality,
'Betweenness Centrality': betweenness_centrality,
'Eigenvector Centrality': eigenvector_centrality)
# 求和并排序
centrality_df['Sum'] = centrality_df.sum(axis=1)
centrality_df = centrality_df.sort_values(by='Sum', ascending=False)
# 保存
centrality_df.to_csv(filepath + 'Ecoli_ppi_centrality.csv')
import numpy as np
import matplotlib.pyplot as plt
from scipy.optimize import curve_fit
# 计算度分布函数
# 即每个度数对应的节点数除以总节点数。
degree_sequence = sorted([d for n, d in G.degree()], reverse=True)
degree_count = np.zeros(max(degree_sequence)+1)
for degree in degree_sequence:
degree_count[degree] += 1
degree_distribution = degree_count/sum(degree_count)
# 绘制度分布函数的双对数轴图
x = np.arange(1, len(degree_distribution)+1)
y = degree_distribution
# plt.loglog(x, y, color = 'chocolate', marker = '.')
plt.plot(x, y, color = 'chocolate', marker = '.')
# 使用幂函数拟合,计算幂指数的值
def power_law(x, a, b):
return a * x ** (b)
popt, pcov = curve_fit(power_law, x, y)
a, b = popt
# plt.loglog(x, power_law(x, a, b), 'c--', label='fit: y=%5.3f*x^(%5.3f)' % (a, b))
plt.plot(x, power_law(x, a, b), 'c--', label='fit: y=%5.3f*x^(%5.3f)' % (a, b))
plt.xlabel('Degree)')
plt.ylabel('Frequency)')
# plt.xlabel('log(Degree)')
# plt.ylabel('log(Frequency)')
plt.legend()
plt.savefig(filepath+'Degree_distribution.png')
plt.show()
print('幂指数的值为:', b)
end_time = time.time()
run_time = end_time - start_time
print("Done (/▽\)")
print("代码运行时间为:分秒".format(int(run_time / 60), round(run_time % 60)))
以上是关于系统生物学大肠杆菌蛋白互作网络分析(系统与合成生物学第三次作业)的主要内容,如果未能解决你的问题,请参考以下文章