chapter1.高通量序列实验简介:设计与生物信息学分析
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了chapter1.高通量序列实验简介:设计与生物信息学分析相关的知识,希望对你有一定的参考价值。
参考技术A2021/4/16
1、设计高通量测序实验的步骤
2、介绍了最广泛使用的应用,并描述了基本的测序概念。
3、可用于生物信息学分析的各种软件程序,以理解测序数据。
1、Insert :用于测序的DNA片段
2、Read : Insert 被测序到的部分
3、Single Read(SR) :一种只从 Insert 序列一端测序的测序程序
4、Pair Read(PR) :一种从 Insert 序列两端测序的测序程序
5、Flowcell :连接DNA芯片并进行测序的一种小玻璃芯片。 Flowcell 被探针覆盖,允许与DNA片段连接的接头杂交。
6、Lane : Flowcell 由8个物理分离的通道组成,称为 Lane 。在所有 Lane 上并行进行测序。
7、Multiplexing/Demultiplexing :在同一 Lane 上对几个样本进行测序称为多路复用 Multiplexing ,在一条 Lane 上测序的 Reads 的分离称为分路复用 Demultiplexing ,通过一个识别每个Reads*的索引将其与已知样本的索引进行比较。
8、Pipeline :一系列的计算过程
(一)reading
1、Resequencing :在一个给定的样本中找到相对于参考基因组的变体
实验细节 :从相关细胞中提取DNA,进行由DNA碎片化和测序组成的样品制备
基本分析总结 :将序列片段映射到参考基因组,并通过总结片段与其基因组位点的差
异来识别相对于参考基因组的变异对应的“地图”
2、Target-enriched sequencing :靶点富集测序是一种特定的 Resequencing 形式,只
关注特定的基因组基因座。
实验细节 :在从细胞中提取DNA并进行样品制备后,进行一个富集过程来捕获相关的
位点,靶富集可以使用“定制的”靶富集探针在基因组的特定区域进行,或
使用可用的试剂盒,如exome-enrichment kits。
基本分析总结 :与 Resequencing 相同
3、De novo assembly :识别一个基因组序列,而无需任何额外的参考
实验细节 :与 Resequencing 相同
基本分析总结 :组装过程依赖于DNA片段的重叠。这些重叠被合并成一致序列,称为
contigs 和 scaffolds 。
(二)counting
1、ChIP-Seq/RIP-Seq :找到RNA或DNA结合蛋白的结合位置
实验细节 :(1)首先,进行了ChIP/RIP实验:蛋白质与DNA/RNA结合,并与之交
联。然后DNA/RNA被分裂。
(2)蛋白质 pull down 经历免疫沉淀过程,交联被逆转
(3)对富集于蛋白结合位点中的DNA/RNA片段进行测序
基本分析总结 :被序列排列的片段被映射到基因组中。基因组中丰富的位置是通过检
测基因组的映射片段的“ peaks (峰)”发现的,这些峰值应该明显高于在
周围的位点中已映射的片段,并且与对照样本相比要高得多----通常
是ChIP实验的输入DNA或其他由非特异性抗体进行的免疫沉淀样
本。
2、RNA-Seq :检测和比较基因表达水平
实验细节 :从细胞中提取总RNA,在样品制备过程中,mRNA被 pull down 并破碎。
然后,mRNA片段被逆转录成cDNA,cDNA片段测序。
基本分析总结 :cDNA片段被映射到参考基因组中。映射到每个基因的片段被计数和
标准化,以便比较不同的基因和不同的样本。在一个RNA-Seq实验
中,通过检测映射到一个未注释区域的基因组上的片段束,可以找到
未标记的基因和转录本。
(三)reading/counting
microRNA-Seq :检测和计数 microRNAs
实验细节 :从细胞中提取总RNA,通过识别大多数已知的microRNA分子共同的自然
结构来分离microRNA,然后对microRNA片段进行逆转录和测序。
基本分析总结 :被测序的片段被映射到基因组中,然后,微RNA可以被检测和计数。
1、在 reading 中,覆盖范围对应于 平均覆盖基因组中每个碱基 的 reads 数量。
一般来说, 30X覆盖率 被认为是识别基因组变异的最小值,而 de novo 通常需要一个更高的覆盖范围。
2、在 counting 中,覆盖的概念并不简单,因为the number of reads along the genome is not expected to be uniform.
可帮助评估是否有足够的reads序列的分析是“ *saturation report* (饱和度报告)”,使用所有的reads确定表达水平,表达水平与 取一部分reads重新计算的表达水平 比较。
1、基因组的重复性
要唯一地对重复区域的read映射进行评分,它必须 比 重复区域 或 边界相邻的非重复序列 更长。更长的reads或PE reads允许“拯救”非唯一端,也映射到基因组中的非唯一区域。
2、差异剪接变异
同一基因表达的转录本不同时:
3、测序样本与参考基因组的遗传距离
如果被测序的样本与参考基因组有遗传距离,可能需要更长的reads来确定基因组中每个read的来源。
4、寻找结构变异
基因组的结构变化,如长的插入或缺失,倒位和易位可以通过Paired-End信息找到。
5、De Novo 装配
挑战:测序错误、低复杂度区域和重复区域等
更长的PE reads会导致更好的装配,使用一些具有不同insert length的序列库可以改进组装过程。
1、 Resequencing:有遗传距离。。。
2、RNA-Seq:使用来自不同重复的数据,并将其合并为一个具有更高统计显著性的值。
3、ChIP-Seq:+控制样本
1、Raw Data 处理
此步骤的可用软件:Illumina’s CASAVA software,Illumina运行会生成“base-calling”文件(*.bcl),它们只有在转换为通用fastq格式时才会非常有用,在此文件转换过程中,还执行解复用过程,即从同一lane上排序的不同样本分离读取。
2、质量控制和read操作
此步骤的可用软件:CASAVA和FastQC
测序运行完成后,在开始分析之前,应检查运行的质量是否以下参数,这些参数可能说明样品和运行的质量。
3、为De Novo Assembly组装 Contigs 和 Scaffolds
此步骤的可用软件:SOAPdenovo,ABySS,Velvet,ALL-PATHS
4、mapping
此步骤的可用软件:BWA ,Bowtie,TopHat
5、 Variant Calling and Filtering
此步骤的可用软件: SAMtools,GATK,MAQ
帮助检测变异的两个基本参数如下:
(1)Coverage at the loci
(2)被测序的等位基因的频率
6、Assembling Transcripts
7、 Gene Expression Analysis
此步骤的可用软件:Cufflinks,Myrna
一种常见的归一化方法FPKM,计算如下:
8、 Peak Detection
此步骤的可用软件:MACS,SICER
测序与芯片高通量数据挖掘与分析学习班
生物信息学(bioinformatics)定义:是在大分子方面的概念型的生物学,并且使用了信息学的技术,这包括了从应用数学、计算机科学以及统计学等学科衍生而来各种方法,并以此在大尺度上来理解和组织与生物大分子相关的信息。
培训目的:
很多老师都做过测序或者芯片实验,虽然测序公司已经给出了一部分的数据分析,比如过滤、质控、比对、定量和差异等,但是这些分析属于基础傻瓜式的标准分析,并不能完整的讲一个故事!因此,需要我们自己掌握高通量数据挖掘的能力,从海量信息中获得自己想要的关键基因!

培训预期:
通过2天的培训,使学员掌握生信文献分析思路和8个实用操作知识点(具体见最后),可以独立完成一篇基于公共数据库的高通量数据挖掘分析。
生物信息学魅力:
帮助预测疾病相关的潜在基因,以及该基因潜在的作用靶点、上游调控转录因子等,从而指导实验方向、缩小试验范围、简化试验流程。
为基金申请提供支持,通过强大的信息数据的收集整理,减少投入增强研究目的性;且通过整合技术优势,指导提高临床诊断水平。
丰富的免费的数据资源和生物软件工具:数据库:NCBI, EBI, DDBJ, PDB,SWISS-PROT等;软件工具:Cytoscape,BLAST, R语言,String, David等。
案例:

该文献的分析流程如下图所示:
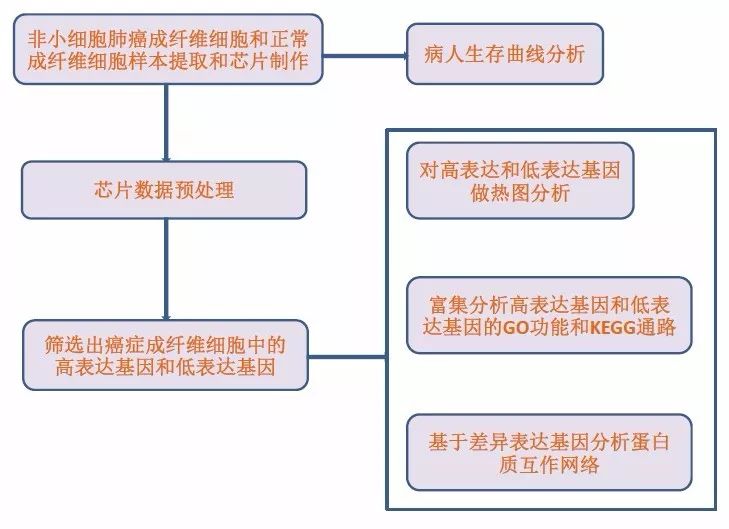
我们可以看到该文献主要的分析步骤有:差异表达分析、聚类热图构建、GO和KEGG 通路分析、蛋白互作网络分析、生存曲线分析等。我们的主题基本上按照类似的文献思路进行讲解,和大家一起分享一下一篇SCI文章的生信分析步骤。
通过2天的学习,我们可以掌握一篇生信SCI文献的所有分析模块,并可以独立的结合自己的研究方向进行生信分析和数据挖掘,获得一套属于自己的分析结果。
第一天上午第一节:基因数据库使用介绍
测序标准报告的解读。
NCBI Gene数据库:最权威的基因综合数据库之一。
NCBI GEO数据库:数据量最大的高通量公共数据库。
GEO2R工具:在线做差异分析的R工具
bioDBnet:在线基因ID转换工具。
Uniprot:最权威的蛋白综合数据库之一。
第一天上午第二节: DAVID工具——功能富集分析
功能富集的原理和意义
基因ID转换
基因功能注释(GO ,KEGG pathway)
基因功能富集(GO ,KEGG pathway)
基因功能聚类(GO ,KEGG pathway)
第一天下午第三节 聚类热图及pathwaymap图
KEGG 数据库:最权威的通路数据库(pathway map 分析)
HEMI软件:构建聚类热图的本地软件
第一天下午第四节. STRING工具——基因/蛋白相互作用分析(PPI)
蛋白互作分析的原理和意义;
单个蛋白的蛋白互作分析;
多个蛋白的蛋白互作分析;
STRING工具的使用步骤:输入、参数设置、输出等。
第二天上午第五节 cytoscape网络做图软件(上)
网络图构建
逐个修改节点的属性和修改边的属性
网络展示方式layout更美观
网络统计分析
第二天下午第六节 cytoscape网络做图软件(下)
批量修改节点的属性和修改边的属性
Cytoscape的常用插件:ClusterONE
Cytoscape的常用插件:BinGO
第二天下午第七节 答疑环节
通过该主题的学习,可以学会:
高通量公共数据的详细信息查找/下载;
在线做差异表达分析
在线blast分析
基因ID转换
通路map图制作
功能富集分析
蛋白互作分析
8.网络图的构建和美化。
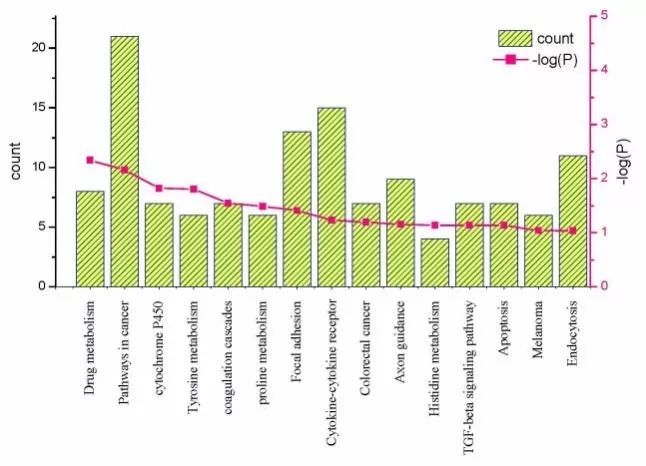
示例图 通路富集分析结果图
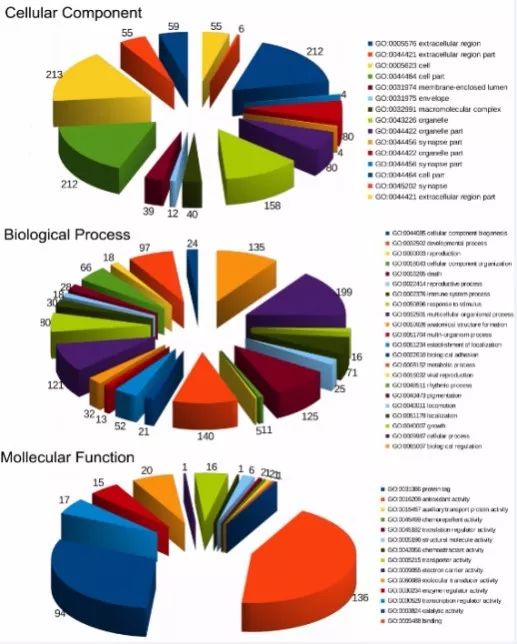
示例图 GO富集分析结果图
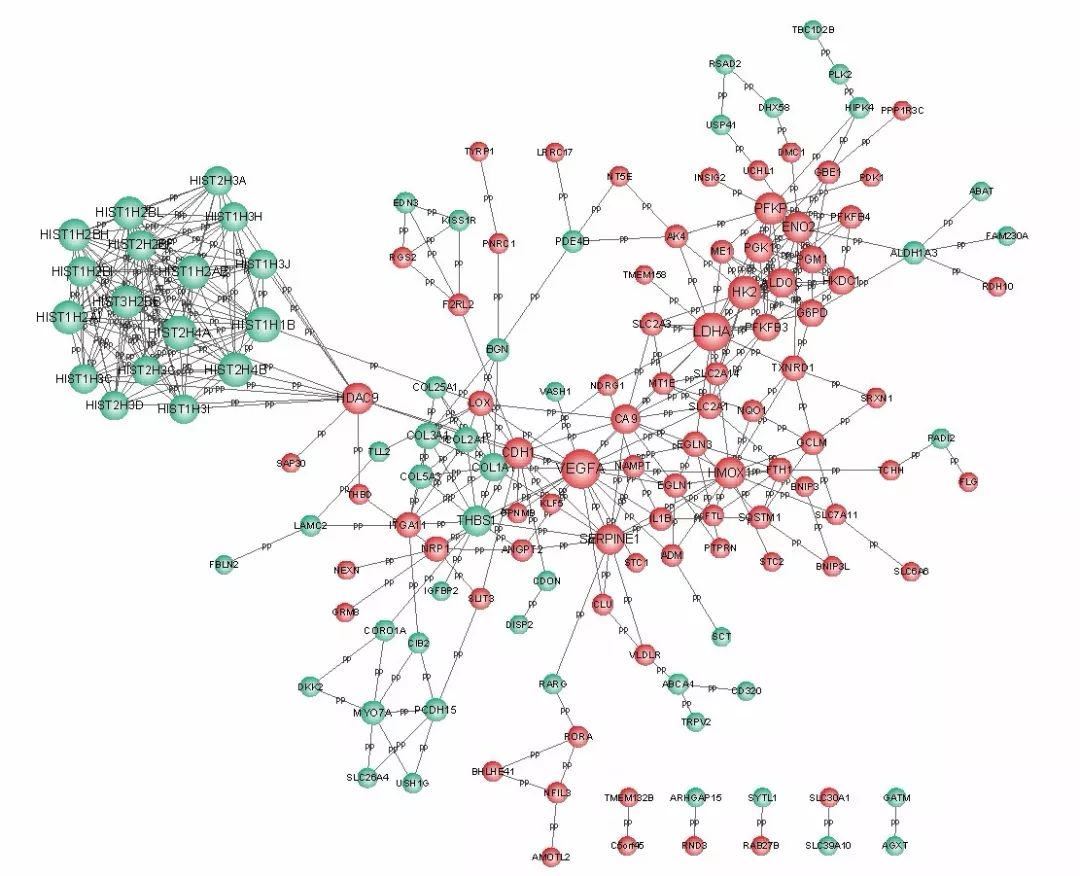
示例图 蛋白互作网络图
示例图 调控网络图
示例图 pathwaymap图
讲师宋博士简介:
成果:参与完成了近百篇软件著作权和发明专利的撰写和申请;肺癌、胰腺癌、骨肉瘤、胃癌等数据库的分析和构建;完成个体基因检测流程和无创唐筛流程的开发。
研究方向:有近十年的生信分析经验,擅长方向有转录组测序分析、芯片数据分析、疾病机理研究分析、疾病预后与基因关联分析、项目分析思路设计以及个性化分析等,精通perl、R等编程语言。
培训经历:在上海、沈阳、济南、武汉等城市举办过十几场培训班。培训的对象有:医生、学生、科研工作者、生信爱好者等。
培训方向:
《测序与芯片数据分析》、《生物信息学的魅力》、《生信文章实例解读》、《生信与实验的密切关系》、《生信与临床医学的关系》、《生信实用工具培训》、《多组学整合分析流程》、《R语言培训》等

学习班时间与地点(上海班)
上海班 培训时间:2018年5月26-27日 (2天)
培训地点: 上海好望角大饭店 上海市肇嘉浜路500号
学习办主办:医药加学习班
学习班承办方:上海遐永医药科技
学习班赞助方:尔云生信
收费标准:
注册费:2500元每位(注册费包含电子版教材、午餐,住宿费自理。)
优惠政策:
1. 提前确认报名及转账的,可以提前拿到学习材料
2. 三人组团报名,每人可优惠100元
3. 四人组团报名,每人收费2300元,
4. 五人组团报名缴费,额外带一人免费注册!
可以开正规会务发票,纸质邀请函(盖红章)。
特别说明:如果需要学习班的上外网路由神器,用于上google学术等国外网站,请自己在邀请函上加900元,我们会在你们的会务费发票上加900元,给您开发票,如果不需要神器,就是按照我们邀请函上的价格,神器可以至少保证用2年以后。
学习班报名方式:
以上是关于chapter1.高通量序列实验简介:设计与生物信息学分析的主要内容,如果未能解决你的问题,请参考以下文章