深度学习构建肿瘤依赖性图谱
Posted Mario cai
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了深度学习构建肿瘤依赖性图谱相关的知识,希望对你有一定的参考价值。
来源于论文
《Predicting and characterizing a cancer dependency map oftumors with deep learning》
代码地址:Code Ocean
大家好呀!今天给大家介绍一篇2021年发表在Science Advances上的文章。
全基因组功能缺失筛查揭示了对癌细胞增殖十分重要的基因,称为肿瘤依赖性。
然而将肿瘤依赖性关系与癌细胞的分子组成联系起来并进一步与肿瘤联系起来还是一个巨大的挑战。
本研究,作者提出了tensorflow框架的深度学习模型Deep—DEP
版本要求:
- tensorflow:1.4.0
- python3.5.2
- cuda8.0.61
- cudnn6.0.21
- h5py==2.7.1
- keras==1.2.2
首先作者队该模型使用无标签的肿瘤基因组(CCL)进行无监督预训练然后保存权重。
无监督预训练(训练集与label一致,带激活函数)
模型流程图:
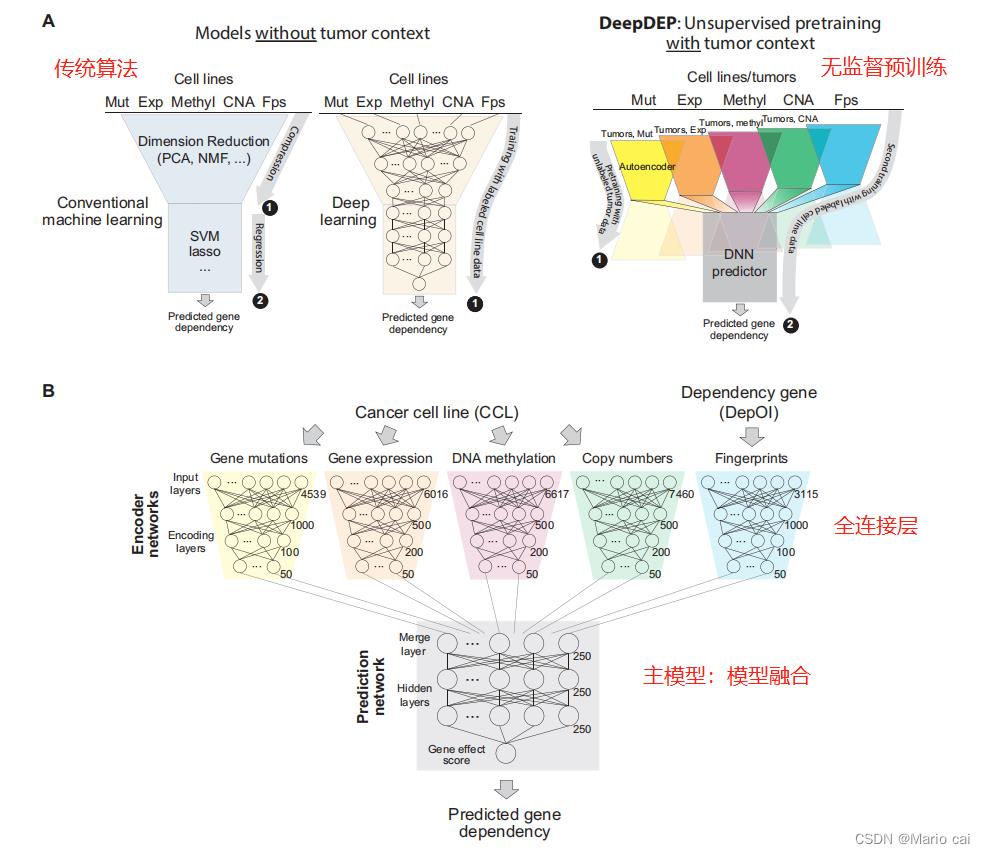
作者使用三个独立数据集验证DeepDEP的性能。通过系统的模型解释,作者扩展了当前的癌症依赖性图谱。将DeepDEP应用于泛癌的肿瘤基因组数据并首次构建了具有临床相关性的泛癌依赖性图谱。总的来说,DeepDEP作为一种新的工具可以用于研究癌症依赖性。
无监督预训练
# Pretrain an autoencoder (AE) of tumor genomics (TCGA) to be used to initialize DeepDEP model training
print("\\n\\nStarting to run PretrainAE.py with a demo example of gene mutation data of 50 TCGA tumors...")
import pickle
from keras import models
from keras.layers import Dense, Merge
from keras.callbacks import EarlyStopping
import numpy as np
import time
def load_data(filename):
data = []
gene_names = []
data_labels = []
lines = open(filename).readlines()#readlines读取全内容
sample_names = lines[0].replace('\\n', '').split('\\t')[1:]#replace将空格替换 #拆分字符串。
dx = 1
for line in lines[dx:]:
values = line.replace('\\n', '').split('\\t')
gene = str.upper(values[0]) #upper将字符串中的小写字母转为大写字母。
gene_names.append(gene)
data.append(values[1:])
data = np.array(data, dtype='float32')
data = np.transpose(data)
return data, data_labels, sample_names, gene_names
def AE_dense_3layers(input_dim, first_layer_dim, second_layer_dim, third_layer_dim, activation_func, init='he_uniform'):
print('input_dim = ', input_dim)
print('first_layer_dim = ', first_layer_dim)
print('second_layer_dim = ', second_layer_dim)
print('third_layer_dim = ', third_layer_dim)
print('init = ', init)
model = models.Sequential()
model.add(Dense(output_dim = first_layer_dim, input_dim = input_dim, activation = activation_func, init = init))
model.add(Dense(output_dim = second_layer_dim, input_dim = first_layer_dim, activation = activation_func, init = init))
model.add(Dense(output_dim = third_layer_dim, input_dim = second_layer_dim, activation = activation_func, init = init))
model.add(Dense(output_dim = second_layer_dim, input_dim = third_layer_dim, activation = activation_func, init = init))
model.add(Dense(output_dim = first_layer_dim, input_dim = second_layer_dim, activation = activation_func, init = init))
model.add(Dense(output_dim = input_dim, input_dim = first_layer_dim, activation = activation_func, init = init))
return model
def save_weight_to_pickle(model, file_name):
print('saving weights')
weight_list = []
for layer in model.layers:
weight_list.append(layer.get_weights())
with open(file_name, 'wb') as handle:
pickle.dump(weight_list, handle)
if __name__ == '__main__':
# load TCGA mutation data, substitute here with other genomics
data_mut_tcga, data_labels_mut_tcga, sample_names_mut_tcga, gene_names_mut_tcga = load_data(r"D:\\DEPOI\\data/tcga_mut_data_paired_with_ccl.txt")
print("\\n\\nDatasets successfully loaded.")
samples_to_predict = np.arange(0, 50)
# predict the first 50 samples for DEMO ONLY, for all samples please substitute 50 by data_mut_tcga.shape[0]
# prediction results of all 8238 TCGA samples can be found in /data/premodel_tcga_*.pickle
print()
input_dim = data_mut_tcga.shape[1]
first_layer_dim = 1000
second_layer_dim = 100
third_layer_dim = 50
batch_size = 64
epoch_size = 100
activation_function = 'relu'
init = 'he_uniform'
model_save_name = "premodel_tcga_mut_%d_%d_%d" % (first_layer_dim, second_layer_dim, third_layer_dim)
t = time.time()
model = AE_dense_3layers(input_dim = input_dim, first_layer_dim = first_layer_dim, second_layer_dim=second_layer_dim, third_layer_dim=third_layer_dim, activation_func=activation_function, init=init)
model.compile(loss = 'mse', optimizer = 'adam')
model.fit(data_mut_tcga[samples_to_predict], data_mut_tcga[samples_to_predict], nb_epoch=epoch_size, batch_size=batch_size, shuffle=True)
cost = model.evaluate(data_mut_tcga[samples_to_predict], data_mut_tcga[samples_to_predict], verbose = 0)
print('\\n\\nAutoencoder training completed in %.1f mins.\\n with testloss:%.4f' % ((time.time()-t)/60, cost))
save_weight_to_pickle(model, r'D:\\DEPOI/results/autoencoders/' + model_save_name + '_demo.pickle')
print("\\nResults saved in /results/autoencoders/%s_demo.pickle\\n\\n" % model_save_name)
经过无监督预训练后,保存权重到pickle文件,以后载入到训练模型上用
主训练
# Train, validate, and test single-, 2-, and full 4-omics DeepDEP models
print("\\n\\nStarting to run TrainNewModel.py with a demo example of 28 CCLs x 1298 DepOIs...")
import pickle
from keras import models
from keras.layers import Dense, Merge
from keras.callbacks import EarlyStopping
import numpy as np
import time
from matplotlib import pyplot as plt
if __name__ == '__main__':
with open(r'D:\\DEPOI/data/ccl_complete_data_278CCL_1298DepOI_360844samples.pickle', 'rb') as f:
data_mut, data_exp, data_cna, data_meth, data_dep, data_fprint = pickle.load(f)
# This pickle file is for DEMO ONLY (containing 28 CCLs x 1298 DepOIs = 36344 samples)!
# First 1298 samples correspond to 1298 DepOIs of the first CCL, and so on.
# For the complete data used in the paper (278 CCLs x 1298 DepOIs = 360844 samples),
# please substitute by 'ccl_complete_data_278CCL_1298DepOI_360844samples.pickle',
# to which a link can be found in README.md
# Load autoencoders of each genomics that were pre-trained using 8238 TCGA samples
# New autoencoders can be pretrained using PretrainAE.py
premodel_mut = pickle.load(open(r'D:\\DEPOI/data/premodel_tcga_mut_1000_100_50.pickle', 'rb'))
premodel_exp = pickle.load(open(r'D:\\DEPOI/data/premodel_tcga_exp_500_200_50.pickle', 'rb'))
premodel_cna = pickle.load(open(r'D:\\DEPOI/data/premodel_tcga_cna_500_200_50.pickle', 'rb'))
premodel_meth = pickle.load(open(r'D:\\DEPOI/data/premodel_tcga_meth_500_200_50.pickle', 'rb'))
print("\\n\\nDatasets successfully loaded.")
activation_func = 'relu' # for all middle layers
activation_func2 = 'linear' # for output layer to output unbounded gene-effect scores
init = 'he_uniform'
dense_layer_dim = 250
batch_size = 10000
num_epoch = 100
num_DepOI = 1298 # 1298 DepOIs as defined in our paper
num_ccl = int(data_mut.shape[0]/num_DepOI)
# 90% CCLs for training/validation, and 10% for testing
id_rand = np.random.permutation(num_ccl)
id_cell_train = id_rand[np.arange(0, round(num_ccl*0.9))]
id_cell_test = id_rand[np.arange(round(num_ccl*0.9), num_ccl)]
# print(id_cell_train)
# prepare sample indices (selected CCLs x 1298 DepOIs)
id_x=np.arange(0, 1298)
id_y=id_cell_train[0]*1298
id_train = np.arange(0, 1298) + id_cell_train[0]*1298
for y in id_cell_train:
id_train = np.union1d(id_train, np.arange(0, 1298) + y*1298)
id_test = np.arange(0, 1298) + id_cell_test[0] * 1298
for y in id_cell_test:
id_test = np.union1d(id_test, np.arange(0, 1298) + y*1298)
print("\\n\\nTraining/validation on %d samples (%d CCLs x %d DepOIs) and testing on %d samples (%d CCLs x %d DepOIs).\\n\\n" % (
len(id_train), len(id_cell_train), num_DepOI, len(id_test), len(id_cell_test), num_DepOI))
# Full 4-omic DeepDEP model, composed of 6 sub-networks:
# model_mut, model_exp, model_cna, model_meth: to learn data embedding of each omics
# model_gene: to learn data embedding of gene fingerprints (involvement of a gene in 3115 functions)
# model_final: to merge the above 5 sub-networks and predict gene-effect scores
t = time.time()
# subnetwork of mutations
model_mut = models.Sequential()
model_mut.add(Dense(output_dim=1000, input_dim=premodel_mut[0][0].shape[0], activation=activation_func,
weights=premodel_mut[0], trainable=True))
model_mut.add(Dense(output_dim=100, input_dim=1000, activation=activation_func, weights=premodel_mut[1],
trainable=True))
model_mut.add(Dense(output_dim=50, input_dim=100, activation=activation_func, weights=premodel_mut[2],
trainable=True))
# subnetwork of expression
model_exp = models.Sequential()
model_exp.add(Dense(output_dim=500, input_dim=premodel_exp[0][0].shape[0], activation=activation_func,
weights=premodel_exp[0], trainable=True))
model_exp.add(Dense(output_dim=200, input_dim=500, activation=activation_func, weights=premodel_exp[1],
trainable=True))
model_exp.add(Dense(output_dim=50, input_dim=200, activation=activation_func, weights=premodel_exp[2],
trainable=True))
# subnetwork of copy number alterations
model_cna = models.Sequential()
model_cna.add(Dense(output_dim=500, input_dim=premodel_cna[0][0].shape[0], activation=activation_func,
weights=premodel_cna[0], trainable=True))
model_cna.add(Dense(output_dim=200, input_dim=500, activation=activation_func, weights=premodel_cna[1],
trainable=True))
model_cna.add(Dense(output_dim=50, input_dim=200, activation=activation_func, weights=premodel_cna[2],
trainable=True))
# subnetwork of DNA methylations
model_meth = models.Sequential()
model_meth.add(Dense(output_dim=500, input_dim=premodel_meth[0][0].shape[0], activation=activation_func,
weights=premodel_meth[0], trainable=True))
model_meth.add(Dense(output_dim=200, input_dim=500, activation=activation_func, weights=premodel_meth[1],
trainable=True))
model_meth.add(Dense(output_dim=50, input_dim=200, activation=activation_func, weights=premodel_meth[2],
trainable=True))
# subnetwork of gene fingerprints
model_gene = models.Sequential()
model_gene.add(Dense(output_dim=1000, input_dim=data_fprint.shape[1], activation=activation_func, init=init,
trainable=True))
model_gene.add(Dense(output_dim=100, input_dim=1000, activation=activation_func, init=init, trainable=True))
model_gene.add(Dense(output_dim=50, input_dim=100, activation=activation_func, init=init, trainable=True))
# prediction network
model_final = models.Sequential()
model_final.add(Merge([model_mut, model_exp, model_cna, model_meth, model_gene], mode='concat'))
model_final.add(Dense(output_dim=dense_layer_dim, input_dim=250, activation=activation_func, init=init,
trainable=True))
model_final.add(Dense(output_dim=dense_layer_dim, input_dim=dense_layer_dim, activation=activation_func, init=init,
trainable=True))
model_final.add(Dense(output_dim=1, input_dim=dense_layer_dim, activation=activation_func2, init=init,
trainable=True))
# training with early stopping with 3 patience
history = EarlyStopping(monitor='val_loss', min_delta=0, patience=100, verbose=0, mode='min')
model_final.compile(loss='mse', optimizer='adam')
model_final.fit(
[data_mut[id_train], data_exp[id_train], data_cna[id_train], data_meth[id_train], data_fprint[id_train]],
data_dep[id_train], nb_epoch=num_epoch, validation_split=1/9, batch_size=batch_size, shuffle=True,
callbacks=[history])
cost_testing = model_final.evaluate(
[data_mut[id_test], data_exp[id_test], data_cna[id_test], data_meth[id_test], data_fprint[id_test]],
data_dep[id_test], verbose=0, batch_size=batch_size)
print("\\n\\nFull DeepDEP model training completed in %.1f mins.\\nloss:%.4f valloss:%.4f testloss:%.4f" % (
(time.time() - t)/60,
history.model.model.history.history['loss'][history.stopped_epoch],
history.model.model.history.history['val_loss'][history.stopped_epoch], cost_testing))
model_final.save(r'D:\\DEPOI\\results_cai/models/model_demo.h5')
print("\\n\\nFull DeepDEP model saved in /results/models/model_demo.h5\\n\\n")
############################################################################################################################
loss = history.model.model.history.history['loss']
val_loss = history.model.model.history.history['val_loss']
fig = plt.figure()
plt.plot(loss, label="Training Loss")
plt.plot(val_loss, label="Validation Loss")
plt.title("Training and Validation Loss")
plt.legend()
fig.savefig("loss.png")
plt.show()预测:观察模型性能
# Predict TCGA (or other new) samples using a trained model
print("\\n\\nStarting to run PredictNewSamples.py with a demo example of 10 TCGA tumors...")
import numpy as np
import pandas as pd
from keras import models
import time
import tensorflow as tf
import pickle
if __name__ == '__main__':
model_name = "model_demo"
model_saved = models.load_model(r"D:\\DEPOI\\results_cai/models/%s.h5" % model_name)#D:\\DEPOI\\results_cai\\models
# model_paper is the full 4-omics DeepDEP model used in the paper
# user can choose from single-omics, 2-omics, or full DeepDEP models from the
# /data/full_results_models_paper/models/ directory
with open(r'D:\\DEPOI/data/ccl_complete_data_28CCL_1298DepOI_36344samples_demo.pickle', 'rb') as f:
data_mut, data_exp, data_cna, data_meth, data_dep, data_fprint = pickle.load(f)
print("\\n\\nDatasets successfully loaded.\\n\\n")
batch_size = 500
# predict the first 10 samples for DEMO ONLY, for all samples please substitute 10 by data_mut_tcga.shape[0]
# prediction results of all 8238 TCGA samples can be found in /data/full_results_models_paper/predictions/
#
# t = time.time()
y = data_dep
data_pred_tmp = model_saved.predict([data_mut,
data_exp,
data_cna,
data_meth,
data_fprint], batch_size=batch_size, verbose=0)
def MSE(y, t):
return np.sum((y - t) ** 2)
T = []
T[:] = y[:, 0]
P = []
P[:] = data_pred_tmp[:,0]
x =(MSE(np.array(P),np.array(T)).sum())
X = x/(data_mut.shape[0])
print(X)以上是关于深度学习构建肿瘤依赖性图谱的主要内容,如果未能解决你的问题,请参考以下文章