怎么用r语言进行生物多样性分析
Posted
tags:
篇首语:本文由小常识网(cha138.com)小编为大家整理,主要介绍了怎么用r语言进行生物多样性分析相关的知识,希望对你有一定的参考价值。
参考技术A #读入数据china <- read.table("F:\\2008年我国其中31个省、市和自治区的农村居民家庭平均每人全年消费性支出.txt",header=TRUE)
distance <- dist(china) #计算距离
china.hc <- hclust(distance) #聚类分析,最长距离法
plot(china.hc, hang = -1) #绘画系谱图
re <- rect.hclust(china.hc, k = 5) #分为5类
re
for (i in 1:5)
print(paste("第",i,"类"))
print(china[re[[i]],]$地区)本回答被提问者采纳
R语言物种丰度矩阵的实现基本操作与生物物种多样性指数计算
本文由“壹伴编辑器”提供技术支持
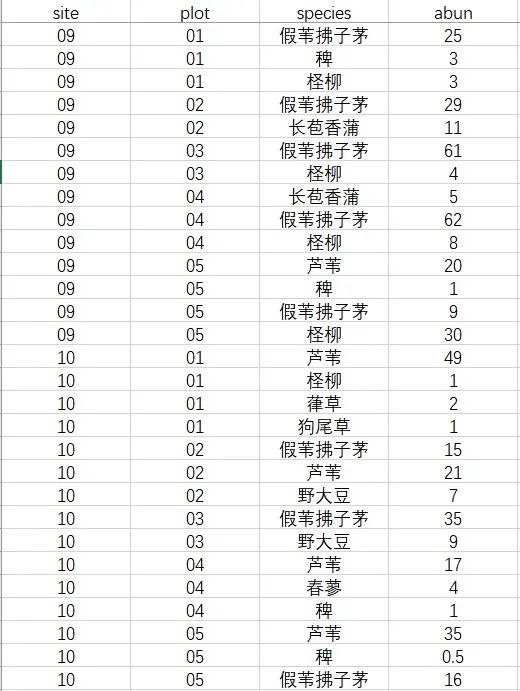
图1 植物群落数据举例
代码如下:
#读取数据
rawdata= read.csv("rawdata.csv", header = T) #第一行为表头
library(doBy)#加载R包(需提前安装)
rawdata.site= summaryBy(abun ~ site + species, data=rawdata, FUN=mean)
#将abun列的数据按照每个样地中的
#每个物种为单位(site +species)
#计算均值
names(rawdata.site)[3]="abun"
#对均值列的列名重新命名为“abun”
head(rawdata.site)#查看前六行的数据
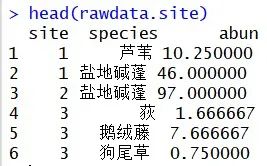
代码如下:
tram.sitedata= function(data1)
{Species=unique(data1$species)
#提取所有的不重复物种名称并赋给
#Species向量
Site=unique(data1$site)
#提取所有不重复样方编号并赋给Site向量
n=length(Species)
#计算不重复物种数并赋值给n
m=length(Site)
#计算不重复的样方数并赋值给m
newdata=as.data.frame(matrix(0,m,n))
#构建一个新m×n的矩阵,然后将矩阵
#转为数据框,才能添加行名和列名
names(newdata)=Species
#将列名改为物种名称
row.names(newdata)=Site
#将行名改为样地的名称
#做一个行循环,然后进行填空
for(iin 1:m)
{newdata1=data1[data1$site==Site[i],]
#提取原始数据第i个样地的数据
newspecies=newdata1$species
#提取第i个样地的物种名
newn=length(newspecies)
#提取第i个样地的物种数
#做一个列循环,看多少个物种,
#就插入多少数值
for (jin 1:newn)
{Ncol=which(Species==newspecies[j])
#提取第i个样地的物种所在的位置
newdata[i,Ncol]=newdata1$abun[j]
#第i行物种所在的位置依次#填入
#当前的物种多度值
}
}
return(newdata)#返回结果
}
spe.matrix= tram.sitedata(rawdata.site)#丰度矩阵
library(vegan)#(需提前安装)
spe.rel.matrix<- decostand(spe.matrix, "total")#相对丰度矩阵
write.csv(spe.rel.matrix,"spe.rel.csv")
#输出结果
将数据读出到spe.rel.csv文件中,结果如下:
图2转换后的相对丰度矩阵
根据物种相对丰度矩阵,我们可以计算生物多样性指数(以物种多样性为例)。
代码如下:
library(vegan)
Richness=rowSums(spe.rel.matrix>0)#物种丰富度指数
Shannon=diversity(spe.rel.matrix,"shannon")
#shannon多样性指数
Simpson=diversity(spe.rel.matrix,"simpson")
#Simpson多样性指数
H=diversity(spe.rel.matrix)
Pielou=H/log(Richness) #Pielou均匀度指数
完
本文编辑
2016级博士 衣世杰
以上是关于怎么用r语言进行生物多样性分析的主要内容,如果未能解决你的问题,请参考以下文章